1) Las neoplasias malignas de las células linfoides van desde leucemias de baja malignidad hasta cánceres invasores y se originan en células del sistema inmune a distintas etapas de diferenciación, lo que da lugar a variedad en morfología y manifestaciones clínicas.
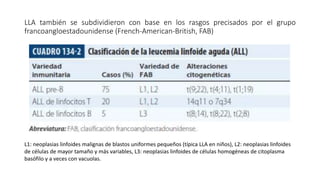
2) La leucemia linfoide aguda es la leucemia más frecuente en niños y se clasifica según la línea linfoide afectada (B o T) y características citogenéticas e inmunológicas.