Este documento resume las características de la leucemia linfática crónica (LLC), incluyendo que afecta principalmente a personas mayores de 55 años, causa linfoadenopatías, esplenomegalia y hepatomegalia, y se diagnostica mediante análisis de sangre periférica y médula ósea que muestran linfocitos pequeños y redondos. Explica los factores pronósticos, sistemas de estadificación y opciones de tratamiento disponibles.







![GAMMAPATÍAS MONOCLONALES
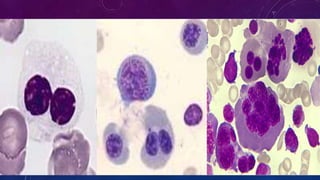
• GM constituyen un grupo de trastornos caracterizados por la proliferación clónal de células linfoides B
en los últimos estadios madurativos
• Incidencia. Muy frecuentes 1% y 5% en personas >50 y 80 años, respectivamente
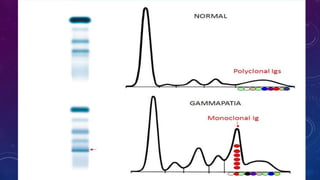
• MGUS—> MMA—> MM
• MGUS no IgM: componente M <3g/dl, plasmocito en MO <10%, sin CRAB
• MGUS IgM: componente M <3g/dl, plasmocito en MO <10%, ausencia de anemia, híper viscosidad,
linfadenopatia, hepatoesplenomegalia.
• MGUS CLL: FLC k/l, aumento de la concentración de CL involucrado, usencia de BM (Ig) por IF.
• MM indolente: componente monoclonal >3g7dl o componente monoclonal urnario >500mg/2hs y/o
infiltración plasmocitaria en MO 10 - 60 % y ausencias de CRAB
• MM: Infiltración. Plasmocitaria en MO >10% o Bx ue confirme plasmocitoma ósea o extramedular.
Baños de órganos blancos (hiperCalcemia serico, insuficiencia Renal, Anemia, lesión Osea) o
Biomarcador de malignidad (CP en MO >60%, Relacion FLC >100, >1 lesión focal en RMN [>5mm])](https://image.slidesharecdn.com/llc-231017233026-fd526540/85/LLC-pptx-8-320.jpg)
![• Estadio:
• Laboratorio
• ECG Ecocardiograma
• BMO( CTF, CTG, FISH, AP, IHQ)
• Estudio de imágenes
• Estadificacion:
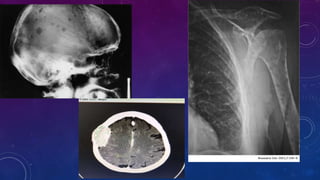
• Durie y Salmon: I; Hb >10g/dl Ca normal , Rx normal o Plasmocitario solitario, IgG <5000mg/dl IgA
<3000mg/dl, Cadenalivianas en orina <4g/24hs. II; no I o III. III; Hb <8,5g/dl, Ca >12mg/dl, esiones osea
avanzado[>4 lesiones osea y/o Fx patologica no vertebral ni costal], IgG >7000mg/dl, IgA >5000g/dl CL en
orina >12g/24hs. A Crea <2mg/dl B >2mg/dl
• ISS: I; B2MG <3,5mg/l y Alb >3,5 mg/dl II; no I oIII. III; B2MG >5,5mg/l
• ISS-R: I; ISS I + LDH Normal, Ausencia de CTG de alto riesgo. II; No I o II. III; ISS III + t(4;14), t(14;16) o del
(17p) o LDH elevado
• Riesgo CTG
• Alto riesgo(15%); t(14;16), t(14;20), del(17p), del 13q, monosomia 13, Cariotipo complejos
• Riesgo intermedio(10%); t(4;14), Ganncia 1q
• Riesgo estandar(75%); Hierdiploidia, t(11;14), t(6;14)](https://image.slidesharecdn.com/llc-231017233026-fd526540/85/LLC-pptx-9-320.jpg)




![• Sinstema pronostico:
• IPSS (Blastos %, CTG, Citopeias)
• IPSS-R (Blastos %, Hb, Plaquetas, Hb, Neutroflos)
• WPSS[WHO2001] (calsificacion WHO,CTG, Requerimiento transfusional)
• MDARSS (CTG, Rec plaquetarios, Hb, PS, GB, Blastos en MO, Requerimiento trasnfucional)
• Tratamiento:
• Soporte trasfucionales
• Eritropoyetina
• G-CSF
• Corticoides
• Lenalidomida
• Androgenos
• Hipometilante
• Ag trombopoyeticos
• Quelante de hierro
• TALLO](https://image.slidesharecdn.com/llc-231017233026-fd526540/85/LLC-pptx-16-320.jpg)