Descargado 322 veces



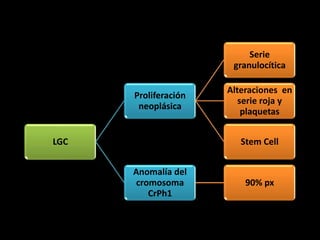

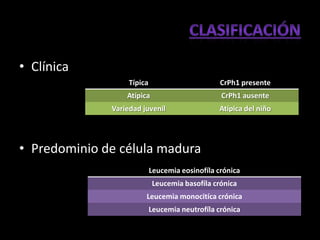






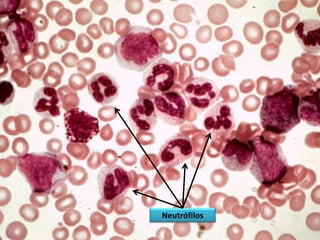

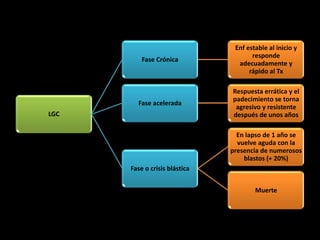












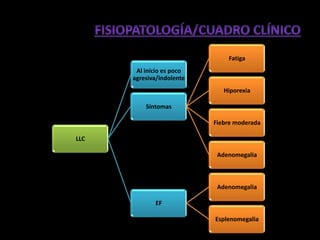




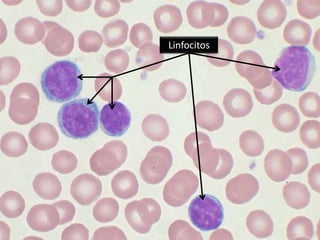












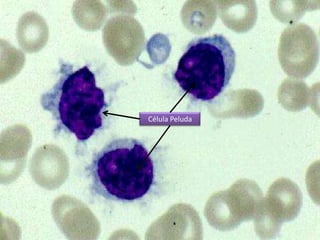





Este documento describe las leucemias crónicas más comunes en México, incluyendo la granulocítica, linfocítica y de células peludas. Estas se caracterizan por un curso indolente y larga evolución sin células muy indiferenciadas, distinguiéndolas de la leucemia aguda. El tratamiento busca controlar los síntomas y mejorar la calidad de vida, aunque rara vez puede lograr la curación excepto mediante trasplante de médula ósea.



































![C:\fakepath\leucemia[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cfakepathleucemia1-100819231029-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)













































