Descargado 165 veces

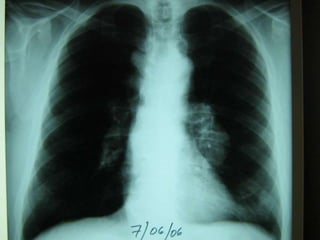



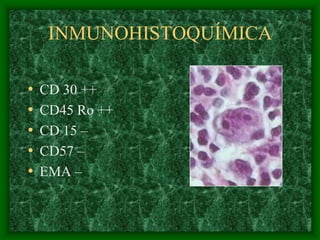

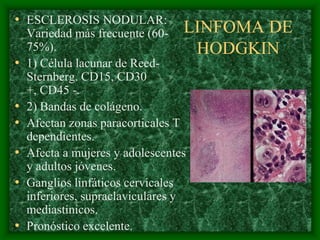
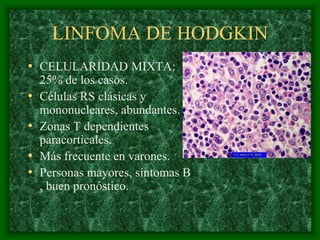































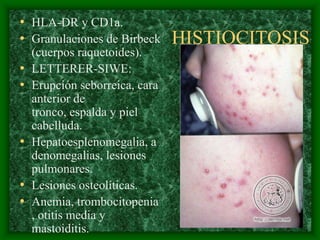



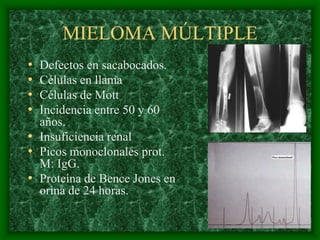


El documento describe las características del linfoma de Hodgkin. Se caracteriza por la presencia de células de Reed-Sternberg en ganglios linfáticos. Existen varias variedades dependiendo del tipo de células presentes. Los síntomas incluyen fiebre, pérdida de peso y agrandamiento ganglionar. El tratamiento depende del estadio y pronóstico suele ser bueno.























































































