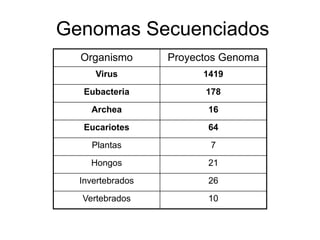
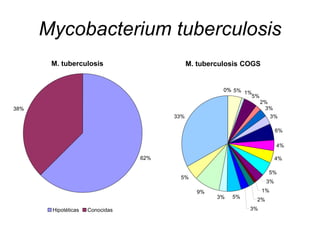
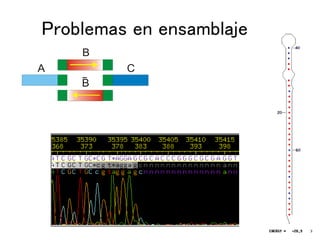
Este documento describe los principales pasos en la secuenciación, limpieza y ensamblaje de genomas. Se han secuenciado miles de genomas de virus, bacterias, arqueas, plantas, hongos e invertebrados y vertebrados. Los pasos incluyen la generación de secuencias, la traducción de cromatogramas, la limpieza de contaminantes y repeticiones, el agrupamiento y el ensamblaje de secuencias usando programas como Phrap. El ensamblaje final se visualiza y edita en Consed para corregir erro