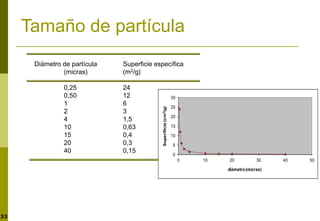
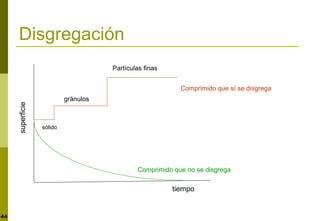
Este documento trata sobre la liberación de fármacos. Explica las diferentes fases de la liberación como la disgregación, disolución y difusión. También describe los factores que afectan a la velocidad de disolución como el pH, la temperatura, la formación de sales y complejos, el tamaño de partícula y la forma cristalina. Finalmente, presenta ensayos para evaluar la cinética de disolución y formas farmacéuticas de liberación modificada.















![16
Disolución y pH
a
o
o
s
K
H
C
C
C
]
[ +
⋅
+
=
La solubilidad de un ácido débil, Cs viene dada por:
Cs = [HA] + [A-]
HA [H+] + [A-]
Ka
[HA]: solubilidad intrínseca de la forma ácida no ionizada (Co)
[A-]: concentración del anión: infinitamente soluble
Para una base débil:
]
[ +
⋅
+
=
H
K
C
C
C a
o
o
s](https://image.slidesharecdn.com/tema3liberacionocw-240201205355-39a03c1e/85/Tema_3_Liberacion_OCW-pdf-16-320.jpg)
![17
Disolución y pH
Sustituyendo en la ecuación de Noyes-Withney:
Para un ácido débil
⋅
+
= +
]
[
'
H
C
K
C
K
dt
dC o
a
o
Para una base débil
⋅
+
=
+
a
o
o
K
H
C
C
K
dt
dC ]
[
'
K’ = D A/h](https://image.slidesharecdn.com/tema3liberacionocw-240201205355-39a03c1e/85/Tema_3_Liberacion_OCW-pdf-17-320.jpg)