
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a 1º 2º 3º ley de la termodinámica
Similar a 1º 2º 3º ley de la termodinámica (20)
Más de rubhendesiderio
Más de rubhendesiderio (20)
Último
Último (20)
1º 2º 3º ley de la termodinámica
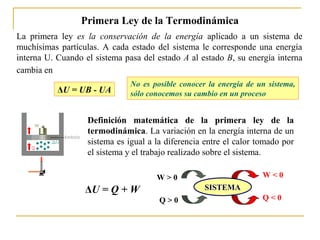
- 1. Primera Ley de la Termodinámica La primera ley es la conservación de la energía aplicado a un sistema de muchísimas partículas. A cada estado del sistema le corresponde una energía interna U. Cuando el sistema pasa del estado A al estado B, su energía interna cambia en Definición matemática de la primera ley de la termodinámica. La variación en la energía interna de un sistema es igual a la diferencia entre el calor tomado por el sistema y el trabajo realizado sobre el sistema. ΔU = Q + W SISTEMA Q > 0 W > 0 W < 0 Q < 0 No es posible conocer la energía de un sistema, sólo conocemos su cambio en un procesoΔU = UB - UA
- 2. ΔU = función de estado extensiva Q y W no son funciones de estado. Ya que conociendo los estados inicial y final no se puede conocer Q o W ∴ Tanto Q como W dependen del camino seguido para ir del estado 1 al estado 2 ΔU = Q + W Ejemplo: Si tomamos 1 mol de H2O a 25 ºC y 1 atm, y elevamos la temperatura hasta 30 ºC, siendo la presión final 1 atm. ¿Cuánto vale Q? No se puede calcular Q, ya que el proceso no está especificado. Se podría aumentar la T, Q = mCpΔT = 18g x 1cal/(g ºC) x 5ºC = 90 cal Pero ¿cómo se eleva la temperatura? U ≡ f (T,P,V)
- 3. Calor y el trabajo se “distinguen” por su efecto sobre las moléculas del entorno Q W • Ambas son formas de variar la energía del sistema • El calor es energía “desordenada” y el trabajo energía “ordenada” • NO son funciones de estado
- 4. ΔU= Q + W Proceso cíclico ΔU = 0 ⇒ W = Q Proceso no es cíclico ΔU ≠ 0 Proceso a volumen constante ΔU = Q Proceso aislado ΔU = W Si el sistema realiza trabajo U disminuye Si se realiza trabajo sobre el sistema U aumenta. Si el sistema absorbe calor al ponerlo en contacto térmico con un foco a temperatura superior, U aumenta Si el sistema cede calor al ponerlo en contacto térmico con un foco a una temperatura inferior, U disminuye. SISTEMA Q > 0 W > 0 W < 0 Q < 0
- 5. • En un sistema adiabático, la energía interna sólo puede cambiar por transferencia de trabajo con el entorno. ΔU = Q + W •En un sistema diatérmico, la energía interna puede cambiar por transferencia de calor y trabajo con el entorno. ΔU = Q + W
- 6. Transformaciones La energía interna U del sistema depende únicamente del estado del sistema, en un gas ideal depende solamente de su temperatura. Mientras que la transferencia de calor o el trabajo mecánico dependen del tipo de transformación o camino seguido para ir del estado inicial al final. - No hay variación de volumen del gas - W=0 - Q=ncV(TB-TA) - Donde cV es el calor específico a volumen constante Proceso Isócorico - ΔU = Q + W = Q - PΔV
- 7. Proceso Isóbarico - No hay variación en la presión del gas - W=p(vB-vA) - Q=ncP(TB-TA) - Donde cP es el calor específico a presión constante -ΔU = Q + W = Q - PΔV
- 8. p V - No hay variación en la temperatura del gas - ΔU = ΔQ - ΔW - Si ΔU = 0 (proceso isotérmico) Entonces 0 = ΔQ - ΔW Por lo tanto, ΔQ = ΔW Proceso Isotérmico Para un gas ideal, la energía interna es independiente del volumen, sólo depende de la temperatura. En consecuencia para cualquier proceso isotermo en un gas ideal ∆U = 0
- 9. Proceso Definición Consecuencia de la 1ra Ley Adiabático Q = 0 ∆U = W Isocórico W = 0 ∆U = Q Cíclico ∆U = 0 Q = W Procesos Específicos y la Primera Ley
- 10. Respuesta: (a) W es el trabajo hecho por el sistema. Es el negativo del trabajo que se hace sobre el sistema. Así que W = - 200J. (b) Q es el calor que entra al sistema. Así que Q = - 70 cal = - 70 * 4.19 J = - 293.3J (c) ∆U = Q + W = -293.3 + 200 = -293.3 + 200 = - 93.3J En un proceso se hacen 200 J de trabajo sobre un sistema durante el cuál se extraen 70 cal de calor. Calcule (a) W, (b) Q y (c) ∆Ucon sus signos.
- 11. • Función de estado H ≡ f (T,P,V,U) • Propiedad extensiva • Unidades de energía (J) • H ≡ H [J/mol] n ENTALPÍA (H) H ≡ U + PV La entalpía es el calor absorbido o liberado en un proceso a presión constante
- 12. PROCESO A PRESIÓN CONSTANTE ΔU = Q + W = Q − ∫ Pext dV = QP - P ∫ dV = QP – P (V2-V1) ΔU=U2-U1 V1 V2 QP = ΔU + P (V2-V1) QP = (U2 + PV2) - (U1+ PV1) = ΔH A presión constante ΔH2 ΔH1
- 13. Reactivos Entalpia(H) Productos ∆H > 0 Reac. endotérmica Entalpia(H) Reactivos Productos ∆H < 0 Reac. exotérmica Exotérmico: calor liberado por el sistema Endotérmico: calor absorbido por el sistema
- 14. Proceso exotérmico es cualquier proceso que cede calor, es decir, transfiere energía térmica hacia los alrededores. Proceso endotérmico, en el cual los alrededores deben suministrar calor al sistema. 2H2 (g) + O2 (g) 2H2O (l) + energía H2O (g) H2O (l) + energía energía + 2HgO (s) 2Hg (l) + O2 (g)
- 15. Relación Qv con Qp (gases) ∆ H = ∆ U + P · ∆ V Aplicando la ecuación de los gases: P · V = n · R · T y si P y T son constantes la ecuación se cumplirá para los estados inicial y final: P · ∆ V = ∆ n · R · T ∆ H = ∆ U + ∆ n · R · T
- 16. Ejemplo: Determinar la variación de energía interna para el proceso de combustión de 1 mol de propano a 25ºC y 1 atm, si la variación de entalpía, en estas condiciones, vale – 2219,8 kJ. C3H8 (g) + 5 O2 (g) → 3 CO2 (g) + 4 H2O (l) ∆ H = –2219,8 kJ Mol reactivos = 1+5 = 6 Mol productos = 3 ⇒∆ n = 3-6=-3 ∆ H = ∆ U + ∆ n · R · T Despejando en ∆ U = ∆ H – ∆ n · R · T = – 2219*103 J + 3 mol · (8,3 J/mol.K) · 298 K = –2214*10+3 J ∆ U = – 2212 kJ
- 17. Relación Qv con Qp (sólidos y líquidos) En reacciones de sólidos y líquidos apenas se produce variación de volumen y ... Qv ≅ Qp es decir: ∆U ≅ ∆H
- 18. Entalpía Normal de la Reacción Es el incremento entálpico de una reacción en la cual, tanto reactivos como productos están en condiciones estándar (P = 1 atm; T = Cte) Se expresa como ∆H0 y como se mide en J o kJ depende de cómo se ajuste la reacción. Así, ∆H0 de la reacción “2 H2 + O2 → 2 H2O” es el doble del de “H2 + ½ O2 → H2O”. ∆H0 = H0 productos – H0 reactivos
- 19. Ecuaciones Termoquímicas Expresan tanto los reactivos como los productos indicando entre paréntesis su estado físico, y a continuación la variación energética expresada como ∆H (habitualmente como ∆H0 ). Ejemplos: CH4(g) + 2 O2(g) → CO2(g) + 2 H2O(l) ∆Hº = –890 kJ H2(g) + ½ O2(g) H2O(g) ∆Hº = –244,4 kJ
- 20. Ecuaciones Termoquímicas ¡CUIDADO!: ∆H depende del número de moles que se forman o producen. Por tanto, si se ajusta poniendo coeficientes dobles, habrá que multiplicar ∆Hº por 2: 2 H2(g) + O2(g) 2 H2O(g) ∆Hº = 2· (–241,4 kJ) Con frecuencia, suelen usarse coeficientes fraccionarios para ajustar las ecuaciones: H2(g) + ½ O2(g) H2O(g) ∆Hº= –241,4 kJ
- 21. H2O (s) H2O (l) Δ H = 6.01 kJ • Los estados físicos de todos los reactivos y productos se deben especificar en las ecuaciones termoquímicas. Ecuaciones Termoquímicas H2O (l) H2O (g) Δ H = 44.0 kJ ¿Cuánto calor se libera cuando 266 g de fósforo blanco (P4) se queman en el aire? P4 (s) + 5O2 (g) P4O10 (s) ΔH = -3013 kJ Respuesta ΔH = - 6470 kJ
- 22. ¿Cuánto calor se emite cuándo una barra de hierro de 869 g se enfría de 94°C a 5°C? Ce de Fe = 0.444 J/g • 0 C Δt = tfinal – tinicial = 50 C – 940 C = -890 C q = mCeΔt = 869 g x 0.444 J/g • 0 C x –890 C = -34339,4 J
- 23. Entalpía estándar de formación (calor de formación). Es el incremento entálpico (∆H) que se produce en la reacción de formación de un mol de un determinado compuesto a partir de los elementos en estado físico normal (en condiciones estándar). Se expresa como ∆Hºf. Se trata de un “calor molar”, es decir, el cociente entre ∆Hº y el número de moles formados de producto. Por tanto, se mide en kJ/mol. Ejemplos: C(s) + O2(g) CO2(g) ∆Hfº = – 393,13 kJ/mol H2(g) + ½ O2(g) → H2O(l) ∆Hfº = – 285,8 kJ/mol
- 24. Cálculo de ∆Hº (calor de reacción) Aplicando la ley de Hess podemos concluir que : ∆Hº = Σ np∆Hºf (productos)– Σ nr∆Hºf(reactivos) Recuerda que ∆Hºf de todos los elementos en estado original es 0.
- 25. Ejemplo: Conocidas las entalpías estándar de formación del butano (C4H10), agua líquida y CO2, cuyos valores son respectivamente –124,7, –285,8 y –393,5 kJ/mol, calcular la entalpía estándar de combustión del butano. La reacción de combustión del butano es: C4H10(g) +13/2O2(g)→ 4 CO2(g) + 5H2O(l) ∆Hºcomb= ? ∆Hº = Σ np∆Hºf(product.) – Σ nr∆Hºf(reactivos) = 4mol(– 393,5 kJ/mol) + 5mol(– 285,8 kJ/mol) – 1mol(– 124,7 kJ/mol) = – 2878,3 kJ/mol Luego la entalpía estándar de combustión será: ∆Hºcombustión = – 2878,3 kJ/mol
- 26. Ley de Hess - Cuando los reactivos se convierten en productos, el cambio en entalpía es el mismo independientemente si la reacción tiene lugar en un paso o en una serie de pasos . - - Recuerda que H es función de estado. - Por tanto, si una ecuación química se puede expresar como combinación lineal de otras, podremos igualmente calcular ∆H de la reacción global combinando las ∆H de cada una de las reacciones.
- 27. Ejemplo: Dadas las reacciones (1) H2(g) + ½ O2(g) H2O(g) ∆Hº1 = – 241,8 kJ (2) H2(g) + ½ O2(g) H2O(l) ∆Hº2 = – 285,8 kJ Calcular la entalpía de vaporización del agua en condiciones estándar. ∆Hºvap= + 44 kJ (1) H2(g) + ½ O2(g) H2O(g) ∆Hº1 = – 241,8 kJ (2) H2O(l) H2(g) + ½ O2(g) ∆Hº2 = + 285,8 kJ H2O(l) H2O(g)
- 28. Esquema de la ley de Hess ∆Hº1 = – 241,8 kJ ∆Hº2 = – 285,8 kJ ∆Hº3 = 44 kJ H H2(g) + ½ O2(g) H2O(g) H2O(l)
- 29. Ejercicio: Conocidas las entalpías estándar de formación del butano (C4H10), agua líquida y CO2, cuyos valores son respectivamente –124,7, – 285,8 y –393,5 kJ/mol, calcular la entalpía estándar de combustión del butano. Si utilizamos la ley de Hess, la reacción: (4) C4H10(g) +13/2O2(g) → 4 CO2(g) + 5H2O(l) ∆H0 comb=? Puede obtenerse a partir de: (1) H2(g) + ½ O2(g) → H2O(l) ∆Hº1 = – 285,8 kJ (2) C(s) + O2(g) → CO2(g) ∆Hº2 = – 393,5 kJ (3) 4C(s) + 5H2(g) → C4H10(g) ∆Hº3 = – 124,7 kJ ΔHºcomb = – 2878,3 kJ
- 30. Ejercicio: Determinar ∆Hºf del eteno (C2H4) a partir de los calores de reacción de las siguientes reacciones químicas: (1) H2(g) + ½ O2(g) → H2O(l) ∆Hº1 = – 285,8 kJ (2) C(s) + O2(g) → CO2(g) ∆Hº2 = – 393,13 kJ (3) C2H4(g) + 3O2(g) → 2CO2(g) + 2 H2O(l) ∆Hº3 = – 1422 kJ ΔHºf = 64,14 kJ
- 31. Ejercicio: Las entalpías de combustión de la glucosa (C6H12O6) y del etanol (C2H5OH) son –2815 kJ/mol y –1372 kJ/mol, respectivamente. Con estos datos determina la energía intercambiada en la fermentación de un mol de glucosa, reacción en la que se produce etanol y CO2. ¿Es exotérmica la reacción? Las reacciones de combustión son, respectivamente: (1) C6H12O6 + 6O2 6CO2 + 6H2O ∆H1 = – 2815 kJ/mol (2) C2H5OH + 3O2 2CO2 + 3H2O ∆H2 = – 1372 kJ/mol ∆H3= – 71 kJ y la reacción es exotérmica.
- 32. Calcule la entalpía estándar de formación de CS2 (l) teniendo que: C(grafito) + O2 (g) CO2 (g) ΔH0 = -393.5 kJ S(rómbico) + O2 (g) SO2 (g) Δ H0 = -296.1 kJ CS2(l) + 3O2 (g) CO2 (g) + 2SO2 (g) Δ H0 = -1072 kJ ΔH0 = 86.3 kJ
- 33. Energía de Enlace “Es la energía necesaria para romper un enlace de un mol de sustancia en estado gaseoso” En el caso de moléculas diatómicas es igual que la energía de disociación: A—B(g) A(g) + B(g) ∆Hdis = Eenlace= Ee Ejemplo: H2(g) 2H(g) ∆H = 436 kJ Es positiva (es necesario aportar energía al sistema) Es difícil de medir. Se suele calcular aplicando la ley de Hess.
- 34. Ejemplo: Calcular la energía del enlace H—Cl en el cloruro de hidrógeno conociendo ∆Hºf (HCl) cuyo valor es –92,3 kJ/mol y las entalpías de disociación del H2 y del Cl2 que son 436,0 kJ/mol y 243,4 kJ/mol, respectivamente. La reacción de disociación del HCl será: HCl(g) H(g) + Cl(g) ∆Hº4= ??????? ½H2(g) + ½Cl2(g) → HCl(g) ∆Hº1 = –92,3 kJ H2(g) 2H(g) ∆Hº2 = 436,0 kJ Cl2(g) 2Cl(g) ∆Hº3 = 243,4 kJ Ee(HCl) = 432,0 kJ/mol
- 35. - Aplicando la ley de Hess en cualquier caso se obtiene la siguiente fórmula: ∆Hº= Σ ni · Ee(enl. rotos) – Σ nj · Ee(enl. formados) en donde ni representa el número de enlaces rotos y formados de cada tipo. Cálculo de ∆Hº a partir de las Energía de Enlace (disociación)
- 36. Ejemplo: Sabiendo que las energía de los siguientes enlaces (kJ/mol): C=C:611; C–C:347; C–H:413 y H–H:436, calcular el valor de ∆Hº de la reacción de hidrogenación del eteno. Reacción: CH2=CH2(g) + H2(g) → CH3–CH3(g) En el proceso se rompe un enlace C=C y otro H–H y se forman 2 enlaces C–H nuevos (el etano tiene 6 mientras que el eteno tenía sólo 4) y un enlace C–C. ∆Hº= Σ Ee(enl. rotos) – Σ Ee(enl. formados) = ∆Hº= 1Ee(C=C) + 1 Ee(H–H) – 1Ee(C–C) – 2 Ee(C–H) ∆ H0 = 1 mol · 611 kJ/mol + 1mol · 436 kJ/mol – (1 mol · 347 kJ/mol + 2 mol · 413 kJ/mol) = ΔHº = –126 kJ/mol
- 37. Enlace Ee (kJ/mol) H–H 436 C–C 347 C=C 620 C≡C 812 O=O 499 Cl–C 243 C–H 413 C–O 315 C=O 745 O–H 460 Cl–H 432 Ejercicio: Calcula el calor de combustión de propano a partir de los datos de energía de enlace de la tabla. C3H8 + 5 O2 → 3 CO2 + 4 H2O Enlaces rotos: 8 C–H, 2 C–C y 5 O=O Enlaces formados: 6 C=O y 8 O–H ∆Hº= Σ Ee(e. rotos) – Σ Ee(e. form.) ∆Hº= 8 Ee(C–H) + 2 Ee(C–C) + 5 Ee(O=O) – [6 Ee(C=O) + 8 Ee(O–H)] ∆Hº= 8·413 kJ + 2·347 kJ +5·499 kJ – (6·745 kJ + 8·460 kJ) = –1657 kJ ∆H0 comb(C3H8) = –1657 kJ/mol
- 38. Entropía (S) Es una medida del desorden del sistema que sí puede medirse y tabularse. ∆S = Sfinal – Sinicial Existen tablas de Sº (entropía molar estándar) de diferentes sustancias. En una reacción química: ∆Sº = Σ np· Sºproductos – Σ nr· Sºreactivos La entropía es una función de estado.
- 39. Segunda Ley de la Termodinámica. “En cualquier proceso espontáneo la entropía total del universo tiende a aumentar siempre” ∆Suniverso = ∆Ssistema + ∆Sentorno ≥ 0 A veces el sistema pierde entropía (se ordena) espontáneamente. En dichos casos el entorno se desordena.
- 40. La entropía (S) es una medida de la aleatoriedad o desorden de un sistema. orden SdesordenS ΔS = Sf - Si Si el cambio de los resultados de inicial a final es un aumento en la aleatoriedad Sf > Si ΔS > 0 Para cualquier sustancia, el estado sólido es más ordenado que el estado líquido y el estado líquido es más ordenado que el estado gaseoso Ssólido< Slliquido<< Sgas H2O (s) H2O (l) ΔS > 0
- 41. Procesos que conducen a un aumento en la entropía (ΔS > 0) Sólido Líquido Líquido Soluto Disolución Disolvente Sistema a T1 Sistema a T2 (T2 > T1 )
- 42. ¿Cómo cambia la entropía de un sistema para cada uno de los procesos siguientes? (a) Condensación de vapor de agua La aleatoriedad disminuye La entropía disminuye (ΔS < 0) (b) Formación de cristales de sacarosa de una disolución sobresaturada La aleatoriedad disminuye La entropía disminuye (ΔS < 0) (c) Calentamiento de hidrógeno gaseoso desde 600 C a 800 C La aleatoriedad aumenta La entropía aumenta (ΔS > 0) (d) Sublimación del hielo seco La aleatoriedad aumenta La entropía aumenta (ΔS > 0)
- 43. Cambios de entropía en el sistema (ΔSsis) aA + bB cC + dD ΔS0 rx dS0 (D)cS0 (C)= [ + ] - bS0 (B)aS0 (A)[ + ] ΔS0 rx nS0 (productos)= S mS0 (reactivos)S- La entropía estándar de una reacción (ΔS0 rx) es el cambio de la entropía para una reacción.
- 44. ¿Cuál es el cambio en la entropía estándar en la siguiente reacción a 250 C? 2CO (g) + O2 (g) 2CO2 (g) S0 (CO) = 197.9 J/K•mol S0 (O2) = 205.0 J/K•mol S0 (CO2) = 213.6 J/K•mol ΔS0 rxn = 2 x S0 (CO2) – [2 x S0 (CO) + S0 (O2)] ΔS0 rxn = 427.2 – [395.8 + 205.0] = -173.6 J/K•mol
- 45. Cambios de entropía en el sistema (ΔSsis) Cuando los gases son producidos (o consumidos) • Si una reacción produce más moléculas de gas que las que consume, ΔS0 > 0. • Si el número total de moléculas disminuye, ΔS0 < 0. • Si ni hay cambio neto en el número total de moléculas de gas , entonces ΔS0 puede ser positivo o negativo PERO ΔS0 será un número pequeño. ¿Cuál es el signo del cambio de la entropía para la reacción siguiente? 2Zn (s) + O2 (g) 2ZnO (s) El número total de moléculas de gas baja , ΔS es negativo .
- 46. Cambios de entropía en los alrededores (ΔSalred) Proceso exotérmico ΔSalred > 0 Proceso endotérmico ΔSalred < 0 Alrededores Alrededores Sistema Sistema Calor Energía Energía
- 47. Ejemplo: Calcula ∆Sº para las siguientes reacciones químicas: a) N2(g) + O2(g) → 2 NO(g); b) 3 H2(g) + N2(g) → 2 NH3(g). Datos: Sº (J·mol–1 ·K–1 ): H2(g) = 130,6; O2(g) =205; N2(g) = 191,5; NO(g) = 210,7; NH3(g) =192,3 ∆Sº = Σ np· Sºproductos – Σ nr· Sºreactivos a) ∆Sº = 2 mol · 210,7 J ·mol–1 ·K–1 –(191,5 J·K–1 + 205 J·K–1 ) =24,9 J·K–1 b) ∆Sº = 2·192,3 J·K–1 – (3 mol ·130,6 J· mol–1 ·K–1 + 191,5 J·K–1 ) =–198,7 J·K–1
- 48. ΔSuniverso = Δ Ssistema + Δ Salrededor > 0Proceso espontáneo: ΔSuniverso = ΔSsistema + ΔSalrededor = 0Proceso en equilibrio:
- 49. Física espontánea y procesos químicos • Una cascada corre cuesta abajo • Un terrón de azúcar se disuelve en una taza de café •El calor fluye de un objeto más caliente a un objeto más frío • Un gas se expande en una bombilla al vacío • El hierro expuesto al oxígeno y agua forma herrumbre espontáneo no espontáneo
- 51. ¿Una disminución en el entalpía significa que una reacción procede espontáneamente? CH4 (g) + 2O2 (g) CO2 (g) + 2H2O (l) ∆H0 = -890.4 kJ H+ (ac) + OH- (ac) H2O (l) ∆H0 = -56.2 kJ H2O (s) H2O (l) ∆H0 = 6.01 kJ NH4NO3 (s) NH4 + (ac) + NO3 - (ac) ∆H0 = 25 kJ H2O Reacciones espontáneas
- 52. “La entropía de cualquier sustancia a 0 K es igual a 0” (máximo orden). Equivale a decir que no se puede bajar de dicha temperatura. ¡CUIDADO! Las S de los elementos en condiciones estándar no son 0 sino que es positiva. Tercera Ley de la Termodinámica
- 53. Energía libre de Gibbs (G) (energía libre o entalpía libre) En procesos a T constante se define como: G = H – T · S ∆G = ∆ H – T · ∆S En condiciones estándar: ∆Gº = ∆Hº – T· ∆Sº ∆Suniverso = ∆Ssistema +∆Sentorno > 0 (p. espontáneos) Multiplicando por “–T” y como “–T ∆Sentorno =∆Hsist” –T · ∆Suniverso = – T · ∆Ssist + ∆Hsist = ∆G < 0 En procesos espontáneos: ∆G < 0 Si ∆G. > 0 la reacción no es espontánea Si ∆G. = 0 el sistema está en equilibrio
- 54. Incremento de energía libre de una reacción (∆G) - G es una función de estado. - Al igual que el incremento entálpico el incremento de energía libre de una reacción puede obtenerse a partir de ∆Gºf de reactivos y productos: ∆Gº = Σ np ∆Gºf (productos) – Σ nr ∆Gºf (reactivos)
- 55. Reactivos Energíalibre(G) Productos ∆G > 0 Energíalibre(G) Reactivos Productos ∆G < 0 T, p = ctes. T, p = ctes. Reac. no espontánea Energía libre y Espontaneidad de las Reacciones Químicas Reac. Espontánea
- 56. No siempre las reacciones exotérmicas son espontáneas. Hay reacciones endotérmicas espontáneas: Evaporación de líquidos. Disolución de sales... Ejemplos de reacciones endotérmicas espontáneas: NH4Cl(s) → NH4 + (aq) + Cl– (aq) ∆ Hº = 14,7 kJ H2O(l) H2O(g) ∆Hº = 44,0 kJ Espontaneidad en las Reacciones Químicas
- 57. Una reacción es espontánea cuando ∆G = ∆H – T ∆S es negativo. Según sean positivos o negativos los valores de ∆H y ∆S (T siempre es positiva) se cumplirá que: ∆ H < 0 y ∆ S > 0 ⇒ ∆G < 0 Espontánea ∆ H > 0 y ∆ S < 0 ⇒ ∆G > 0 No espontánea ∆ H < 0 y ∆ S < 0 ⇒ ∆G < 0 a T bajas ⇒ ∆G > 0 a T altas ∆ H > 0 y ∆ S > 0 ⇒ ∆G < 0 a T altas ⇒ ∆G > 0 a T bajas Espontaneidad en las Reacciones Químicas
- 58. ∆H > 0 ∆S > 0 Espontánea a temperaturas altas ∆H < 0 ∆S > 0 Espontánea a todas las temperaturas ∆H < 0 ∆S < 0 Espontánea a temperaturas bajas ∆H > 0 ∆S < 0 No Espontánea a cualquier temperaturas ∆H ∆S Espontaneidad en las Reacciones Químicas
- 59. Ejemplo: ¿Será o no espontánea la siguiente reacción en condiciones estándar? 2H2O2(l)→ 2H2O (l) + O2(g) Datos: ∆Hºf (kJ/mol) H2O(l) = –285,8; H2O2(l) = –187,8 ; Sº (J/·mol K) H2O(l) = 69,9; H2O2(l) = 109,6; O2(g) =205,0. ∆Hº= Σ np∆Hfº(productos)– Σ nr∆Hfº(reactivos) = ∆Hº= 2 ∆Hfº(H2O) + ∆Hfº(O2) – 2 ∆Hfº(H2O2) = 2 mol(–285,8 kJ/mol) – 2 mol(–187,8 kJ/mol) = –196,0 kJ ∆Sº = Σ np· Sºproductos – Σ nr· Sºreactivos = ∆Sº = 2Sº(H2O) + Sº(O2) – 2Sº(H2O2) = 2 mol(69,9 J/mol·K) + 1 mol(205, J/mol·K) – 2mol(109,6 J/mol·K) = 126,0 J / K = 0,126 kJ / K ∆Gº = ∆Hº – T · ∆ Sº= –196,0 kJ – 298 K · 0,126 kJ/ K = ∆Gº = – 233,5 kJ luego será espontánea
- 61. 5.- 6.- 7.- 8.-
- 62. 9.-
- 63. 10.-
- 64. 11.-
Notas del editor
- Ejercicios 1 a 5 (página 60)