El documento describe las características químicas del agua y los solutos, incluyendo su función como disolvente biológico principal y su importancia en la regulación del pH. También describe los carbohidratos, incluyendo los monosacáridos como la glucosa y fructosa, los disacáridos como la lactosa y sacarosa formados por la unión de monosacáridos, y las funciones de los carbohidratos en el cuerpo.


![2
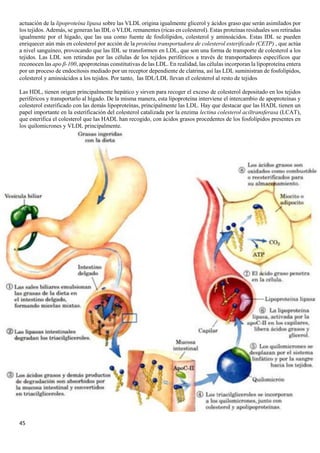
Por tanto, el número de protones va a ser igual al número de iones hidronio (véase en la tabla)
El pH de una disolución es una medida de concentración de los protones; como los protones reaccionan con el agua para
dar iones hidronio, se puede considerar el pH como la concentración de esta última especie química. Debido a que el
pH solo es una manera de expresar la concentración del ion hidronio, las disoluciones acidas y básicas a una temperatura
de 25 grados, pueden identificarse por los siguientes valores de pH:
[H+] pH= - log [H+]
Disoluciones neutras 10-7
M 7
Disoluciones ácidas >10-7
M <7 (menor que 7)
Disoluciones básicas <10-7
M >7 (mayor que 7)
❖ El pH de los fluidos corporales es 6.5 - 8
Ciertos grupos funcionales presentes en las moléculas biológicas pueden comportarse como ácidos o bases débiles. Por
ello su estado de ionización dependerá de la concentración de protones del medio. Si se tiene en cuenta que la mayoría
de las enzimas van a presentar este tipo de grupos ionizables en su centro activo, se comprenderá el importante papel
que puede jugar una pequeña fluctuación del pH celular. Por ejemplo, si el grupo amino de una enzima presenta carga
positiva a un pH 7, un ligero aumento del pH puede forzar a que el H+
del grupo amino sea cedido al medio y, por lo
tanto, pierda esa carga. Con la carga perdida la enzima dejará de funcionar.
Tanto en el medio intracelular como en el extracelular, será por tanto imprescindible una regulación del pH para que las
moléculas puedan operar de manera óptima. Los tampones sistemas acuosos que tienden a amortiguar los cambios que
se producen en el pH, cuando se añaden pequeñas cantidades de ácido (H+
) o de base (OH-
). Estos sistemas de tampón
están constituidos por un ácido débil y su base conjugada, o bien por una base débil y su ácido conjugado. Cuando la
concentración de ambas especies es similar, entonces el sistema tiene una capacidad amortiguadora. En esta situación,
cualquier aumento de la concentración de H+
podrá ser absorbida por la base conjugada, y si se incrementa la
concentración de OH-
, será el ácido débil del sistema tampón quien ceda un portón al medio que neutralice el ion
hidroxilo. Según la ecuación de Henderson cuando el valor de pH de la solución es igual al pKa del sistema, entonces
las concentraciones de las dos especies que definen el sistema de tapón serán iguales. Por lo tanto, en la célula aquellas
sustancias que tengan un pKa próximo a 7 (pH fisiológico) serán buenos tampones.
El principal tampón biológico intracelular es el tampón fosfato, que presenta un pKa de 6,86 de manera que es capaz
de resistir los cambios de pH entre 5,9 y 7,9.](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-2-320.jpg)













![18
3.6. CINÉTICA ENZIMÁTICA
La cinética enzimática es el análisis cuantitativo del efecto de cada uno de los factores que intervienen en la actividad
enzimática, que se evalúa a través de la velocidad de la reacción catalizada. Las variables más importantes son:
• Concentración de enzima, sustratos y productos (incluyendo inhibidores y/o activadores).
• pH_ los cambios en el pH del medio pueden alterar al estado de ionización de las cadenas laterales de
aminoácidos ácidos y básicos. Estos cambios pueden alterar a la afinidad de la enzima por el sustrato, si se ven
alteradas las cargas de los aminoácidos que participan en la formación de interacciones no covalentes en el
complejo ES.
• Temperatura_ el aumento de la temperatura conduce a un aumento en la velocidad de la reacción. Sin embargo,
cuando se sobrepasa el limite de temperatura la enzima se desnaturaliza. Aproximadamente 45 grados es el
valor óptimo para que las enzimas, y otras proteínas, puedan llevar a cabo su función sin llegar a
desnaturalizarse.
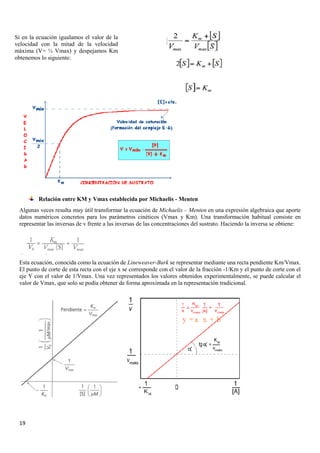
3.6.1 Cinética de las reacciones de un solo sustrato
La variación de la velocidad de una reacción enzimática en función de la concentración del sustrato viene dada por la
ecuación de Michaelis - Menten.
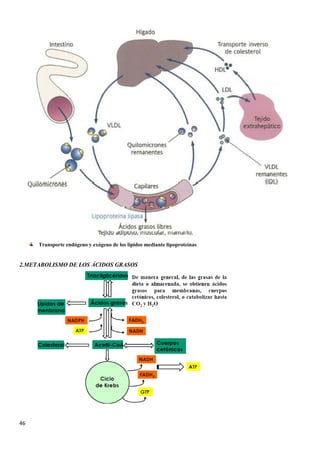
Para la mayoría de las reacciones catalizadas por enzimas, los
datos experimentales proporcionan gráficas, como la de
arriba, en las que se identifican tres zonas claramente
diferenciadas:
• Una primera etapa lineal, abajas concentraciones de
sustrato, en la que la reacción se comporta como si
fuera de primer orden, en la cual la velocidad de
reacción depende únicamente del sustrato.
• Una segunda etapa curvilínea, a concentraciones de
sustrato intermedias, en las que hay un descenso en la
respuesta al aumento de la concentración de sustrato.
• Una última etapa, a elevadas concentraciones de
sustrato en las que la velocidad no varía al aumentar
la concentración de sustrato. La reacción sigue una
cinética de orden cero, donde la velocidad es
independiente de la concentración del sustrato, y se
dice que hay efecto de saturación de la enzima por el
sustrato.
Donde:
• V es la velocidad de la reacción para una determinada concentración de
sustrato [S].
• Vmax es la velocidad máxima de la reacción
• Km es una constante denominada constante de Michaelis-Menten,
característica de cada enzima.](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-18-320.jpg)






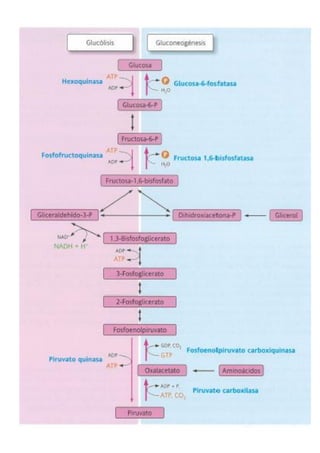

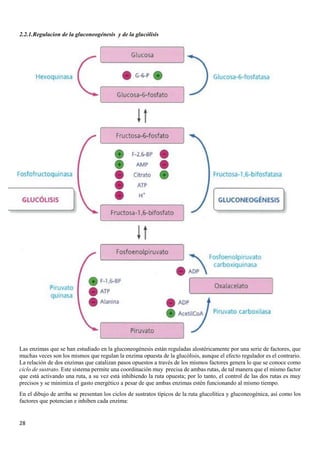
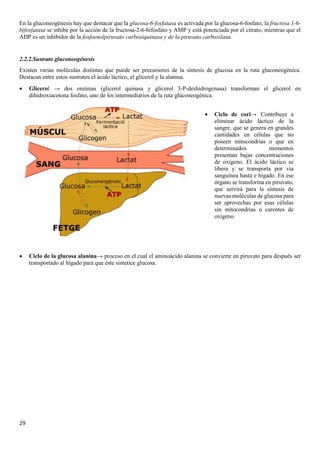

![31
❖ Fermentación alcohólica (una descarboxilación no oxidativa del piruvato)
Este tipo de fermentación se da sobre todo en levaduras y en algunos tipos de bacterias y está implicada en la
elaboración de bebidas alcohólicas y en la fabricación de pan. La fermentación alcohólica realmente consiste
en dos reacciones consecutivas:
o La primera implica la descarboxilación del piruvato por la enzima piruvato descarboxilasa
o La segunda reacción produce etanol a partir de los productos de la reacción anterior por acción de la
alcohol deshidrogenasa.
2.3.2. Descarboxilación oxidativa del piruvato
El piruvato todavía tiene bastante energía química y se puede ser empleada para obtener una cantidad sustancia de ATP.
Para ello el piruvato debe oxidarse por completo. El primer paso es una descarboxilación que origina una molécula de
acetil-CoA, la cual posteriormente se degradará en el llamado ciclo de Krebs, produciendo mucha energía. Este proceso
de eliminación de un átomo de carbono se conoce como descarboxilación oxidativa del piruvato y lo realiza un complejo
METABOLISMO DEL ALCOHOL
Cualquier sustancia del cuerpo ajena es un cenobítico (fármaco, alcohol…). Todos siguen la misma ruta de
detoxicación en el hígado. Dentro de los hepatocitos el metabolismo del alcohol se realiza en tres lugares distintos:
- En citosol por la ADH (alcohol deshidrogenasa). En esta vía el etanol se oxida en acetaldehído por acción de la
alcohol deshidrogenasa, dando lugar poder reductor (NADH). Luego el acetaldehído se oxida en ácido acético
por acción de aldehído deshidrogenasa, dando lugar a poder reductor (NADH). El acetaldehído esta mas oxidado
que el etanol mientras que el ácido acético está más oxidado que el acetaldehído.
- En el peroxisoma por la catalasa
- En el sistema membranoso de oxidación de alcohol se da en el retículo endoplasmático. Aquí se tendría la ruta
del citocromo P 450. Este mismo sistema de oxido-reducciones se utiliza para detoxicar muchos fármacos y está
muy activo en los alcohólicos. Aquí se dan las mismas conversiones en el citosol, solo que se libera O2 y se
catalizan por dos enzimas diferentes (oxidasa de función mixta) y aldehído oxidasa.
La acumulación de NADH favorece fermentación (aumenta la [lactato] y disminuye pH de sangre) e inhibe la PDH
(piruvato deshidrogenasa), favoreciéndose anabolismo a partir de piruvato (rutas anapleróticas) y acetato.
La vitamina B12, acelera el metabolismo del alcohol y a la larga restaura el estado de oxidación de la PDH](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-31-320.jpg)
![32
multienzimático, conocido como piruvato deshidrogenasa (PDH), compuesto por 5 coenzimas y tres enzimas con
actividades enzimáticas distintas:
1. Piruvato descarboxilasa→ produce la eliminación del átomo de carbono del piruvato. Para actuar requiere
pirofosfato de tiamina como cofactor y origina como producto acetaldehído.
2. Dihidrolipoil transacetilasa → emplea dos lipoamidas como cofactores enzimáticos, que utiliza para transferir
el acetaldehído a una molécula de coenzima A (CoA-SH), a la vez que lo oxida originado el acetil CoA. La
reacción que cataliza la conversión de piruvato a acetil-CoA es irreversible y es un importante punto de control
de todo el metabolismo.
3. Dihidrolipoil deshidrogenasa→ esta enzima ayuda en la regeneración de las lipoamidas de la dihidrolipoil
transacetilasa, para la cual requiere como cofactor FAD, que las oxida. Finalmente, los electrones de la
oxidación serán cedidos por el FADH2 a una molécula de NAD+
, formando NADH+H+.
El balance de la reacción del complejo del piruvato deshidrogenasa sería el siguiente:
El complejo de piruvato deshidrogenasa se encuentra fuertemente regulado, presenta una regulación alostérica basada
principalmente en la relación NADH+H+
/NAD+
y acetil CoA/ CoA-SH; de tal manera que
- Si está relación es elevada, lo cual indica un nivel energético alto de la célula, la enzima se inhibe
- Si la relación es baja, lo cual indica bajo nivel energético de la célula, la enzima se activa.
También presenta un control por fosforilación-desfosforilación (modificación covalente reversible) que afecta a la
primera enzima o actividad enzimática (la piruvato descarboxilasa) del complejo piruvato deshidrogenasa.
- Las altas [NADH] y [acetil-CoA] activan la piruvato deshidrogenasa quinasa (PDHK) que fosforila la PDH,
inactivándola.
- Las elevadas [NAD+] y [CoA] inhiben a la piruvato deshidrogenasa quinasa (PDHK) y activan a la piruvato
deshidrogenasa fosfatasa (PDHPP) que desfosforila la PDH, activándola.
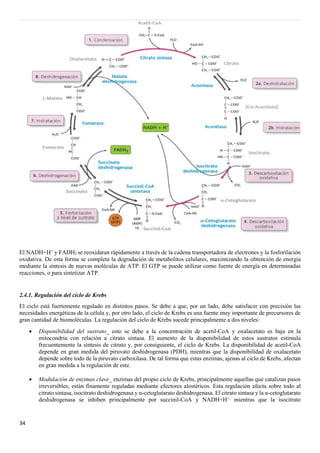
2.4. CICLO DE KREBS
También llamado ciclo del ácido cítrico o ciclo de los ácidos tricarboxílicos. Tiene lugar en la matriz mitocondrial de
la célula, excepto la succinil-CoA sintetasa que forma parte del complejo II de la cadena de transporte de electrones. Es
parte de la vía catabólica que realiza la oxidación de las moléculas de acetil-CoA hasta producir CO2, liberando gran
cantidad de energía química en forma de GTP y de poder reductor (NADH+H+
y el FADH2). En el ciclo de Krebs se
produce una secuencia de reacciones que oxidan los dos átomos de carbono de una molécula de acetil CoA hasta rendir
CO2. Las reacciones son:
1. Condensación entre una molécula de acetil- CoA (dedos carbonos) y una molécula de oxalacetato (de cuatro
carbonos). Esta reacción es una condensación aldólica a la que sigue una hidrolisis que libera la coenzima A
libre. Dicha reacción está catalizada por el citrato sintasa, dando como producto final el citrato (de seis
carbonos). Es un paso irreversible.
2. Transformación del citrato en isocitrato. Con esta reacción que sucede en dos pasos, una deshidratación
seguida de una hidratación se pasa de un sustrato con un alcohol terciario a una molécula con un alcohol
secundario, que resulta más fácilmente oxidable. Esta reacción se lleva a cabo por la aconitasa, formando un
intermediario conocido como cisaconitato.
3. Primera descarboxilación oxidativa. Se produce la transformación del isocitrato (de seis carbonos) a α-
cetoglutarato (de cinco carbonos) reacción que conlleva la reducción de una molécula de NAD+
a NADH+H+
y
la eliminación de un átomo de carbono en forma de CO2. Esta reacción la cataliza el isocitrato deshidrogenasa
y es la primera etapa en la se produce NADH+H+
y CO2.Es un paso irreversible.](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-32-320.jpg)



![38
cuando se produce un ejercicio inmensos. Para la degradación del glucógeno en glucosa 1-P se necesitan
fundamentalmente :
• Glucógeno fosforilasa→ elimina las moléculas de glucosa de los extremos no reductores, recortando las
cadenas lineales de glucógeno. Esta enzima también rompe los enlaces (α1→4), liberando moléculas de glucosa-
1-fosfato.
• Enzima desramificante→ elimina los puntos de ramificación.
Después por acción de la fosfoglucomutasa, la glucosa-1-P se transforma en glucosa-6-P. En el tejido muscular, la
glucosa-6-fosfato se oxidará en la glucolisis. En el tejido hepático será transformada en glucosa libre por la glucosa-6-
6-fosfatasa, y posteriormente se liberará al torrente circulatorio para elevar los niveles de glucosa en sangre en otros
tejidos-
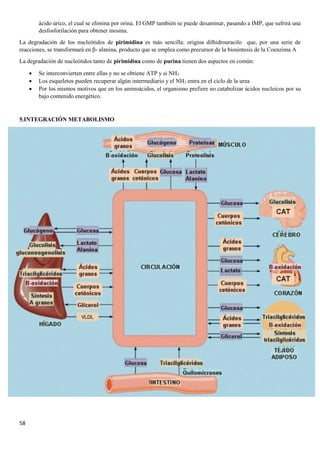
3.3.REGULACIÓN HORMONAL DEL METABOLISMO DEL GLUCÓGENO
La síntesis y la degradación del glucógeno están reguladas cuidadosamente para que la disponibilidad de glucosa permite
cubrir las necesidades energéticas del organismo. El control de estos procesos metabólicos se produce principalmente a
través de tres hormonas: la insulina, el glucagón y la adrenalina.
• Glucagón→ Naturaleza peptídica y síntesis en páncreas. Actúa únicamente en el hígado, puesto que el músculo
no tiene receptores para el glucagón Se libera cuando la [glucosa] en sangre es baja y desencadena rutas para la
obtención de energía a partir de materia de reserva, por lo que:
- En hígado activa la glucogenólisis y la gluconeogénesis
• Insulina → Naturaleza peptídica y síntesis en páncreas. Se libera cuando la [glucosa] en sangre es alta y
desencadena rutas biosintéticas para poder almacenar material de reserva, por lo que
- En hígado y músculo activa la glucogenogénesis.
- En el hígado activa la glucólisis.
• Adrenalina→ Es una catecolamina que se sintetiza en glándulas suprarrenales y se produce rápidamente ante
una respuesta de peligro para dotar al organismo de energía necesaria para la huida por lo que en el músculo
activa la glucogenólisis y la glucólisis.](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-38-320.jpg)

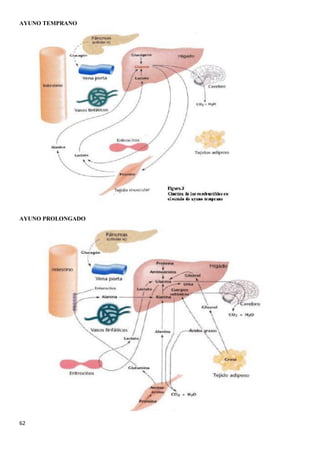
![40
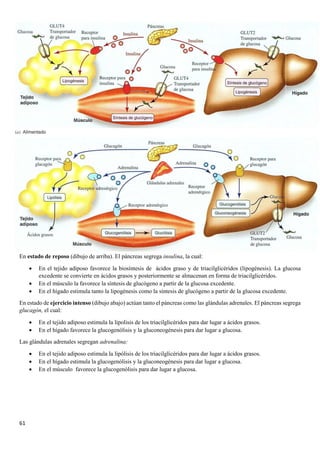
3.4.BIOQUÍMICA DE LA DIABETES
Al no haber insulina no se capta la glucosa por los
tejidos, al no estar estos permeabilizados, por lo
que la [glucosa] aumenta en sangre.
Las células al no entrar en la sangre las células del
cuerpo comienzan a utilizar las grasas del tejido
adiposo para obtener energía. Los ácidos grasos
van a parar a la sangre y luego al hígado, donde se
convierten en acetil- CoA,
Mientras en el músculo se degradan proteínas al no
haber casi glucógeno , lo que produce la liberación
de aminoácidos en sangre. Los aminoácidos van al
hígado, ahí se convierten en glucosa para
convertirse después en ácidos grasos y
posteriormente en acetil CoA
El piruvato, y en este caso, los ácidos grasos
producen acetil-CoA que, además de utilizarse
junto con el oxalacetato en el ciclo de Krebs, es un
precursor de los cuerpos cetónicos. Los cuerpos
cetónicos se producen cuando no hay suficiente
oxalacetato como para que el acetil-CoA reaccione
con él, no pudiéndose iniciar el ciclo de Krebs . La
degradación de los ácidos grasos produce mucha
más aceltil-CoA que el piruvato .](https://image.slidesharecdn.com/apuntesbioquimicapdf-181210170306/85/Apuntes-bioquimica-pdf-40-320.jpg)