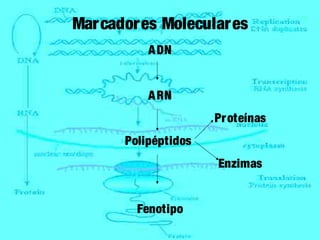
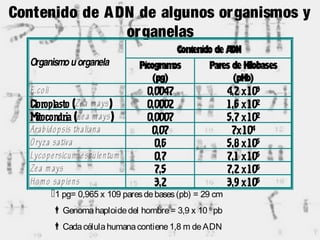
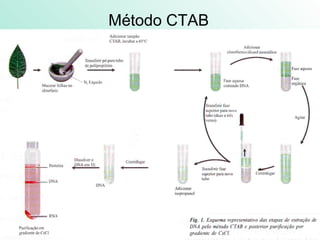
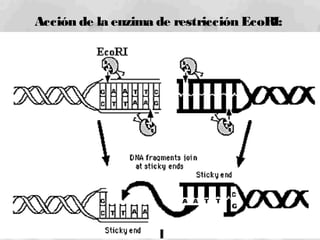
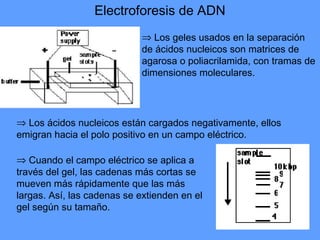
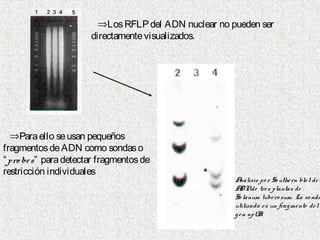
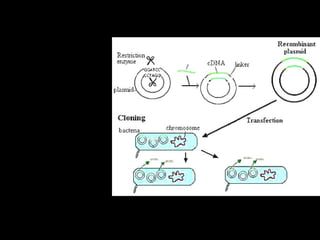
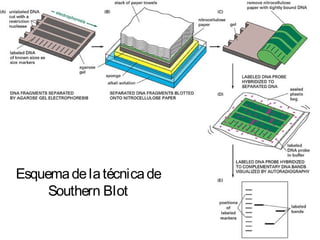
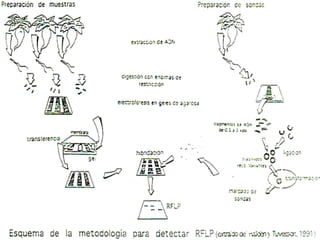
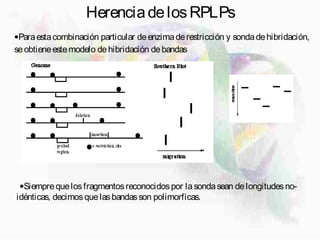
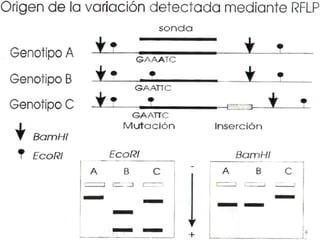
1) Los marcadores moleculares incluyen ADN, ARN, proteínas y enzimas que se utilizan para identificar variedades y caracterizar organismos. 2) Los marcadores de ADN como RFLP se basan en la digestión con enzimas de restricción y detección de polimorfismos en los fragmentos mediante hibridación con sondas. 3) Las aplicaciones de los marcadores moleculares incluyen mapeo genético, selección asistida por marcadores y backcross avanzado.