Descargado 155 veces








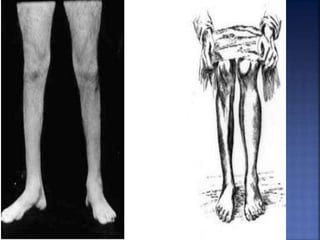


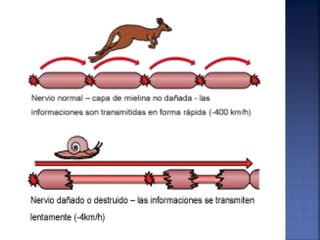

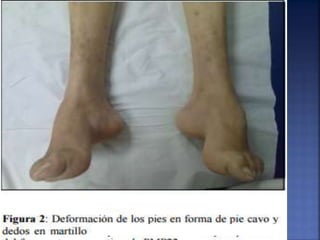



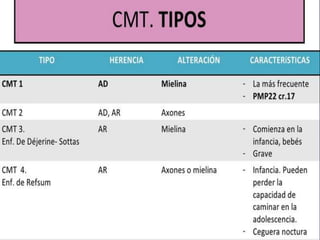

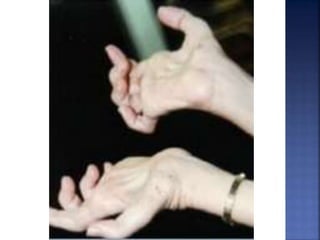

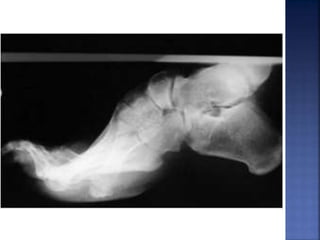

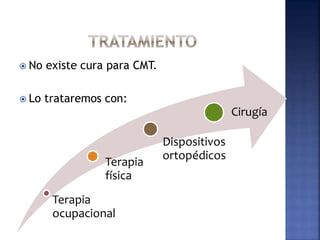
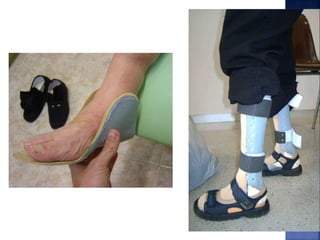
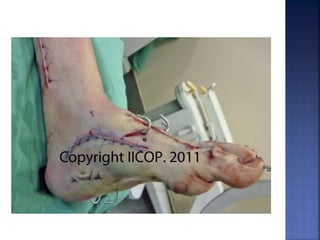




La enfermedad de Charcot-Marie-Tooth (CMT) es una neuropatía hereditaria que afecta los nervios periféricos y se manifiesta en debilidad muscular, deformidades en los pies y pérdida de sensibilidad, afectando aproximadamente a 1 de cada 2,500 personas. Existen varios tipos de CMT, que varían en gravedad y síntomas, y la enfermedad es causada por mutaciones genéticas heredadas de los padres. No hay cura, pero se pueden manejar los síntomas a través de terapia ocupacional, fisioterapia y dispositivos ortopédicos.



































































