Descargado 127 veces










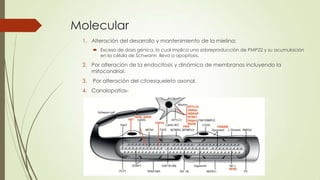
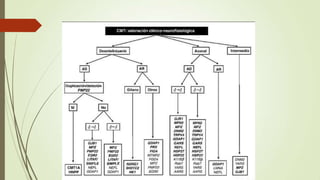








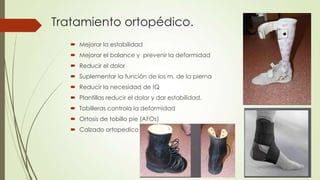



Este caso clínico describe a un paciente de 34 años con una historia familiar de trastornos de la marcha y síntomas de debilidad y dolor en las piernas que sugieren una neuropatía sensitivomotora hereditaria periférica. Los exámenes electrofisiológicos y el estudio genético confirmaron el diagnóstico de enfermedad de Charcot-Marie-Tooth, una enfermedad hereditaria de las neuronas motoras y sensoriales periféricas. El tratamiento se centró en la rehabilitación, el soporte ortop












































![Fraturas dos membros_superiores[1]](https://cdn.slidesharecdn.com/ss_thumbnails/fraturasdosmembrossuperiores1-140305134800-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)










































