Descargado 42 veces































































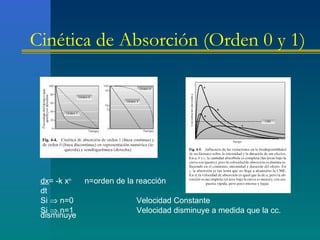
![Absorción
Componentes de la cinética de primer orden:
A) Una fase de absorción [A]
B) El tiempo máximo [Tmax] en que se logra la concentración máxima,
el cual es independiente de la dosis
C) La concentración máxima [Cmax], la cual se obtiene cuando el
fármaco absorbido es igual al fármaco eliminado
D) El segmento de la curva que declina más rápidamente a partir de la
Cmax, el cual representa el proceso de distribución [D]
E) El segmento de la curva que declina más lentamente, a prtir del
punto de inflexión del segmento anterior, el cual representa la
eliminación definitiva del fármaco, a expensas de los procesos de
excreción y biotransformación
F) El área bajo la curva [ABC] que representa la biodisponibilidad](https://image.slidesharecdn.com/fkinetic-130719125355-phpapp02/85/Fkinetic-65-320.jpg)








































El documento describe las diferentes formas farmacéuticas para la administración de medicamentos, incluyendo sólidos, semisólidos, líquidos y gaseosos. Explica los distintos tipos como polvos, tabletas, cápsulas, soluciones, suspensiones, parches y formas para administración oral, rectal, intravenosa, intramuscular y otras vías. El objetivo es elaborar preparados que faciliten la dosificación y administración de principios activos de manera segura y eficaz.



































































