Descargado 312 veces


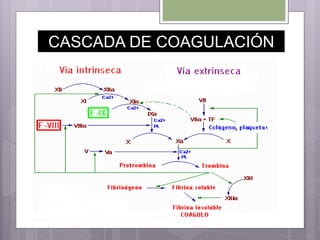

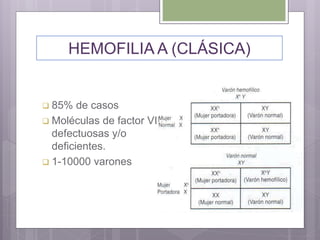










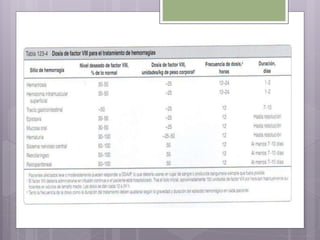







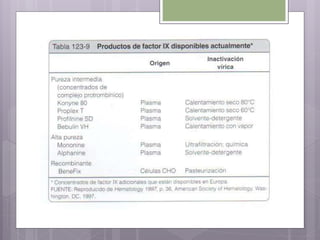






Este documento resume los tres tipos principales de hemofilia: hemofilia A, hemofilia B y hemofilia C. La hemofilia A es la más común y se debe a defectos en el factor VIII. La hemofilia B se debe a defectos en el factor IX. La hemofilia C implica defectos en el factor XI. Todas conducen a un sangrado prolongado debido a defectos en la cascada de coagulación de la sangre. Se describen las características genéticas, clínicas y de tratamiento de cada tipo.



































































