Descargado 26 veces




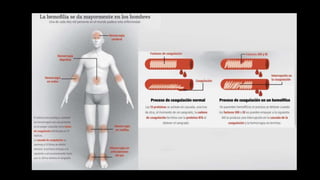

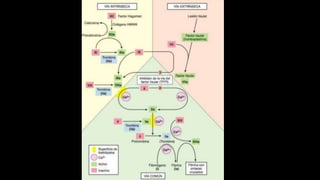
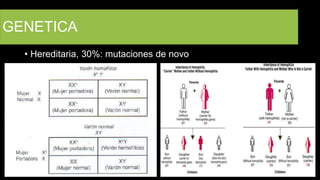
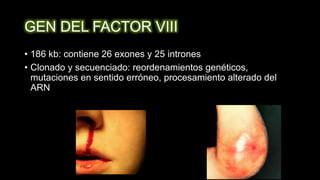

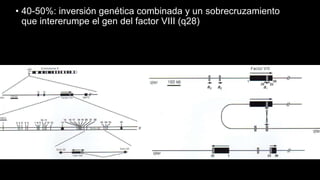
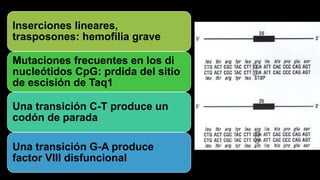

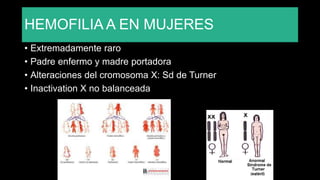


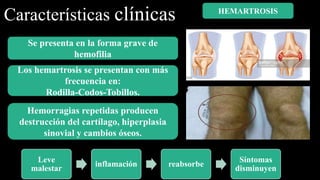
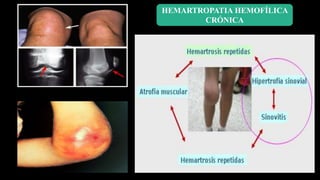
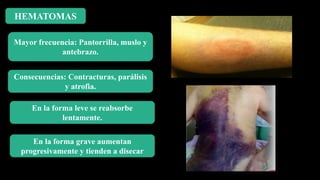
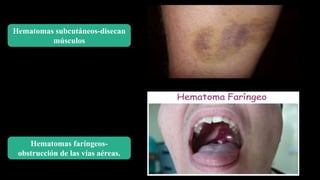




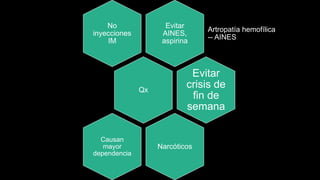
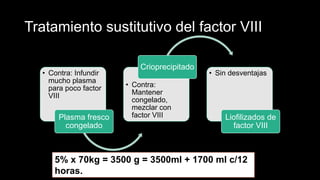
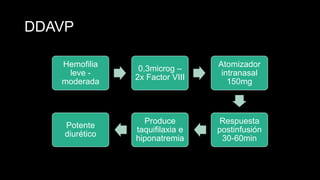

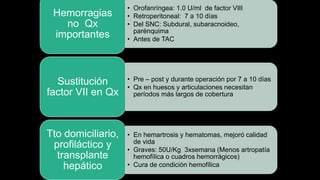

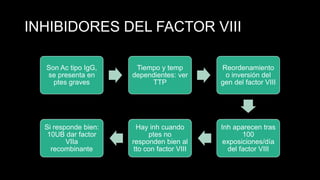


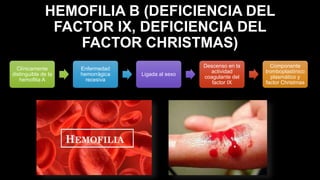
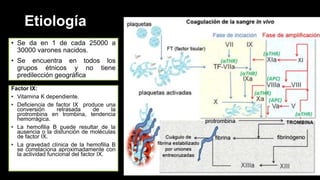
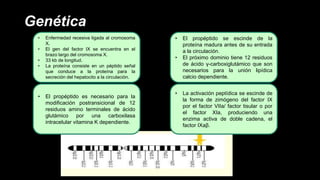
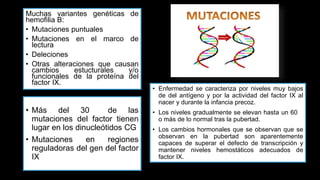
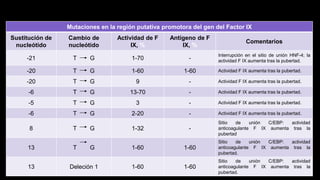
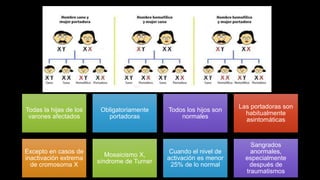








Este documento describe la hemofilia A y B. La hemofilia A es causada por deficiencia del factor VIII y afecta a uno de cada 15,000 habitantes en Ecuador. La hemofilia B es causada por deficiencia del factor IX y afecta a uno de cada 25,000-30,000 varones. Ambas son enfermedades hemorrágicas hereditarias ligadas al sexo que se manifiestan clínicamente con hemartrosis y hematomas. El tratamiento incluye factores de reemplazo y medidas para prevenir hemorragias.





























































































