Descargar para leer sin conexión





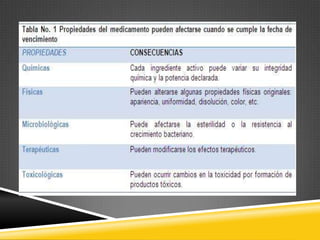



























El documento trata sobre la estabilidad de los medicamentos. Explica que la estabilidad se define como la capacidad de un producto farmacéutico de mantener sus características físicas, químicas y microbiológicas dentro de límites aceptables durante su periodo de validez. Se realizan diferentes tipos de estudios de estabilidad para determinar este periodo, incluyendo estudios acelerados y a largo plazo, y se especifican los análisis requeridos para diferentes formulaciones farmacéuticas.






















































