Descargado 115 veces
















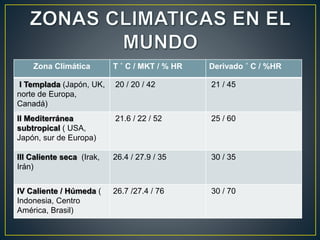






Este documento trata sobre la estabilidad de las formas farmacéuticas. Explica que la estabilidad se refiere a la integridad física y química de la unidad de dosificación y su capacidad de mantener la protección contra la contaminación microbiológica. También cubre factores que afectan la estabilidad como la temperatura, humedad y componentes del envase. Además, detalla los diferentes tipos de pruebas de estabilidad y parámetros a evaluar para cada forma farmacéutica.

























































































