Descargar para leer sin conexión











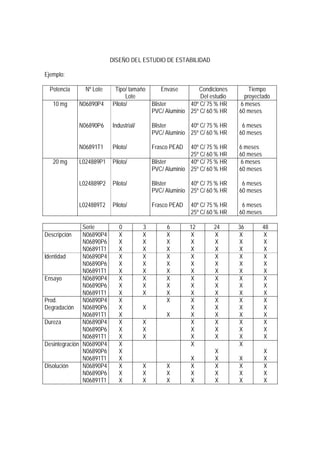





La resolución aprueba una guía para la realización y presentación de estudios de estabilidad de productos farmacéuticos en Chile. La guía estandariza los requisitos mínimos para los estudios de estabilidad y uniforma conceptos y criterios relacionados. La guía debe ser cumplida por la industria farmacéutica para obtener registro sanitario o modificar productos ya registrados.















































