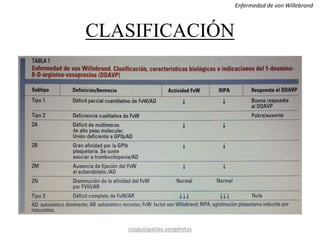
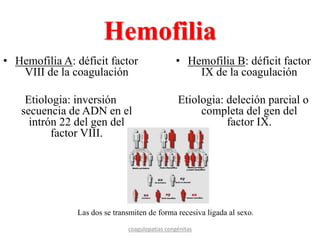
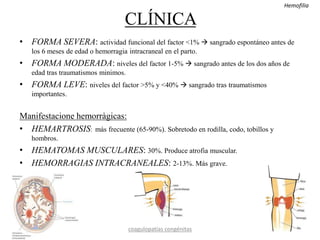
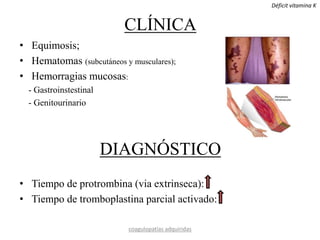
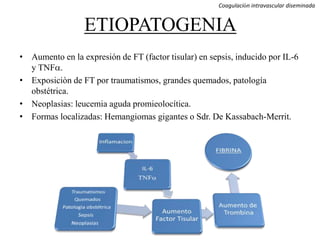
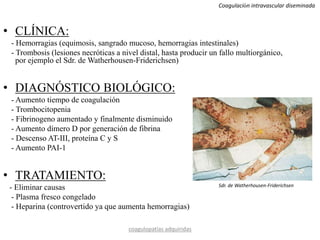
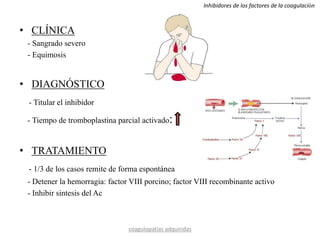
Este documento resume diferentes tipos de coagulopatías, incluyendo coagulopatías congénitas como la enfermedad de von Willebrand, hemofilia A y B, y coagulopatías adquiridas como el déficit de vitamina K, coagulación intravascular diseminada, inhibidores de factores de coagulación, y trastornos de la coagulación relacionados con hepatopatía crónica. Describe las causas, manifestaciones clínicas, diagnóstico y tratamiento de estas diferentes alteraciones de la coagulación.











![• DIAGNÓSTICO DE LABORATORIO
- Descenso tiempos de coagulación (TP t TTPA)
- Disminucion [proteínas C, S antitrombina III] en plasma
- Trombocitopenia
- Aumento dímero D
• TRATAMIENTO
- Desmopresina (acorta tiempo de hemorragia en pacientes con cirrosis)
- Vitamina K (10 mg al día durante 3 días)
- Plasma fresco concentrado (para corregir el TP prolongado)
- Antifibrinolíticos: ácido tranexámico y aminocaproico (en caso de extracciones
dentarias)
- Complejo protrombínico
coagulopatías adquiridas
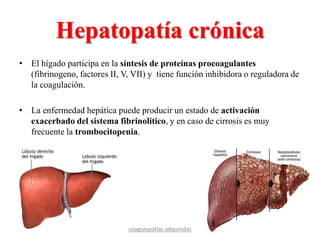
Hepatopatía crónica](https://image.slidesharecdn.com/trabajocoagulopatias-140416065843-phpapp01/85/COAGULOPATIAS-21-320.jpg)















































![Coagulopatias adquiridas[2]](https://cdn.slidesharecdn.com/ss_thumbnails/coagulopatiasadquiridas2-130202130647-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)








































