Descargado 45 veces

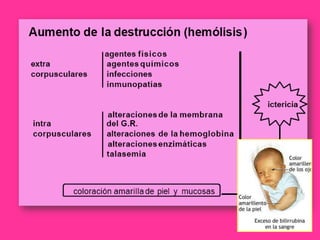

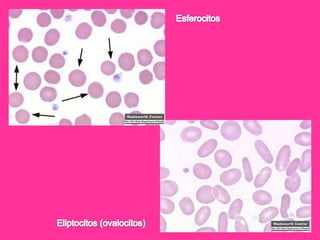
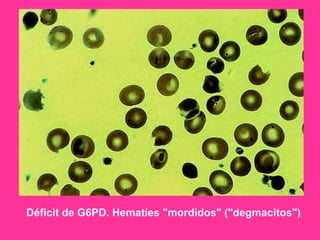

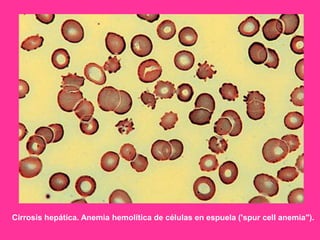

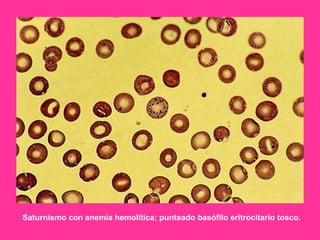
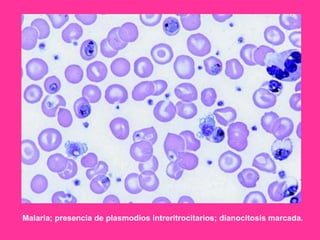
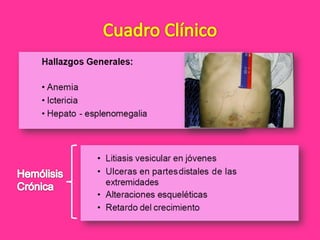



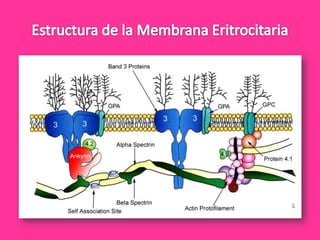
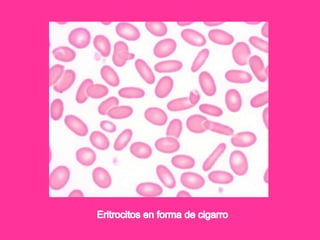


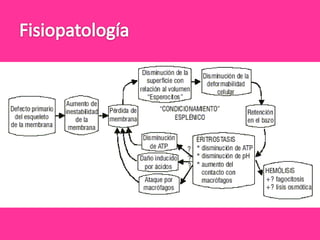

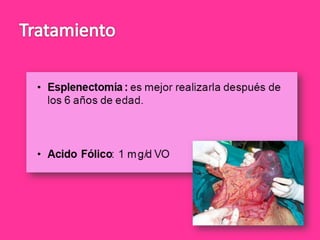



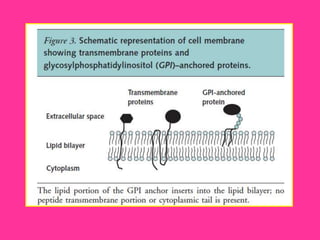

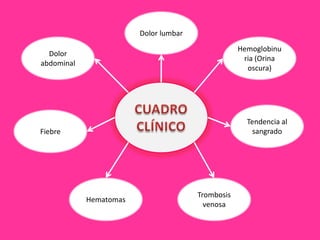
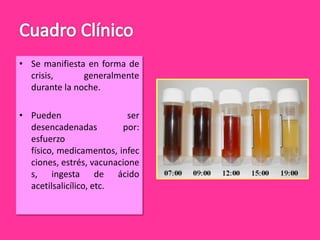





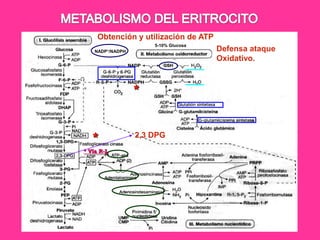




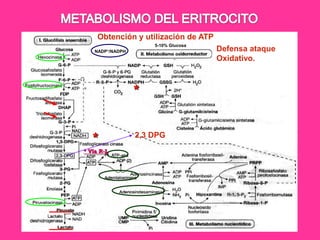

El documento describe varios tipos de anemias hemolíticas, incluyendo déficit de G6PD, esferocitosis hereditaria, hemoglobinuria paroxística nocturna, y enzimopatías del metabolismo del glutatión y de la glicólisis anaeróbica. Estas condiciones se caracterizan por daño a los eritrocitos que resulta en su destrucción prematura y anemia. Algunas son hereditarias mientras que otras pueden ser adquiridas. Los síntomas incluyen fatiga, dolor abdominal y tendencia al























































































