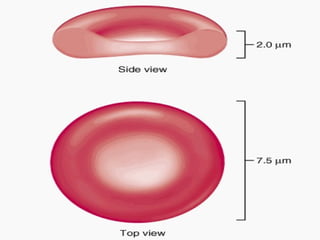
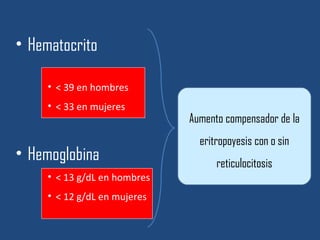
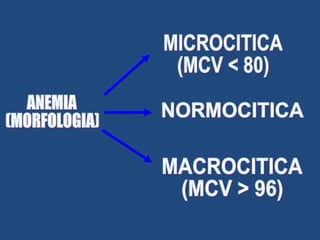
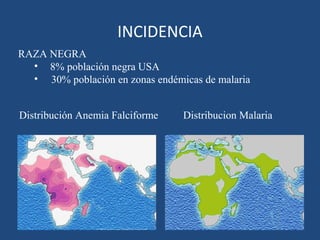
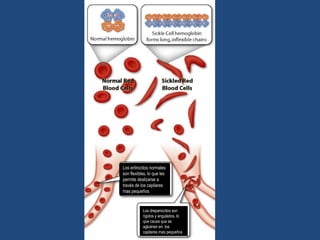
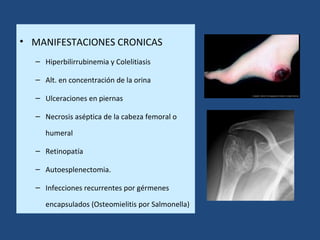
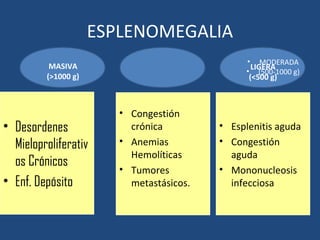
Este documento resume diferentes tipos de anemias, incluyendo sus causas, síntomas, morfología y tratamiento. Describe anemias como la ferropénica, megaloblástica, hemolítica, drepanocítica y de origen inmune. También cubre patologías de bazo y timo como esplenomegalia y timoma.