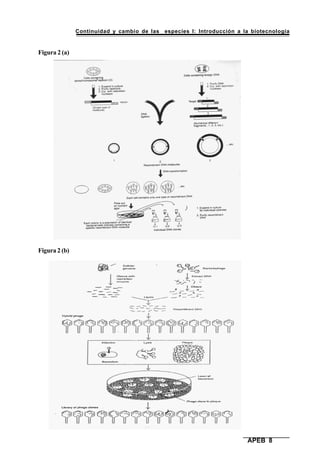
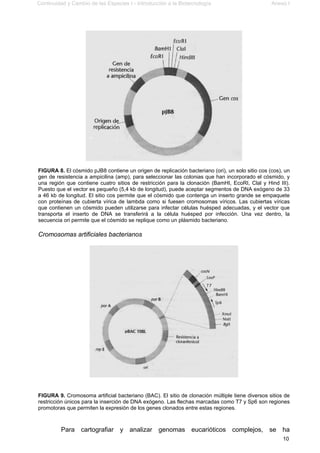
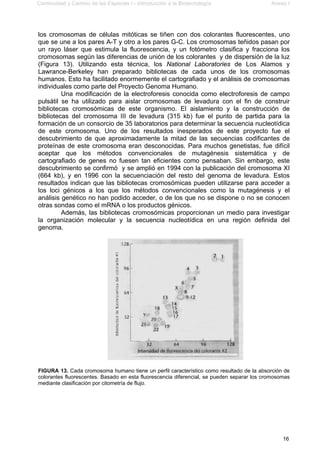
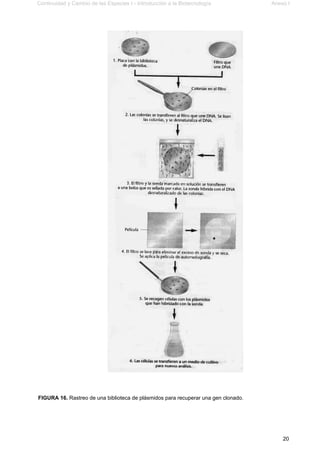
Este documento presenta el segundo módulo de un curso sobre introducción a la biotecnología. El módulo se enfoca en técnicas de amplificación molecular de células, incluyendo estrategias básicas como el clonado in vivo y el clonado in vitro. También describe diferentes tipos de vectores como plásmidos, fagos y cromosomas artificiales de levadura, y cómo se pueden utilizar para clonar DNA. El documento incluye actividades prácticas para que los estudiantes apliquen estos concept