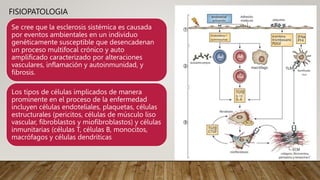
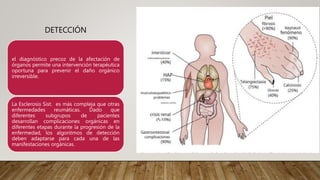
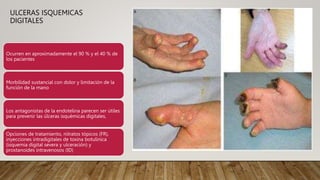
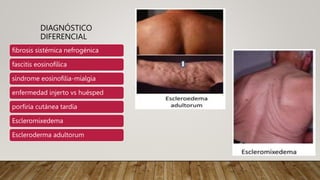
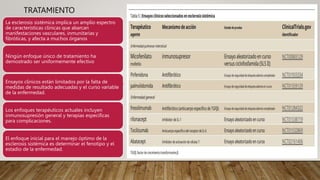
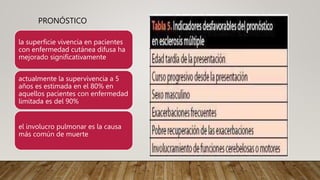
La esclerosis sistémica es una enfermedad autoinmune caracterizada por daño vascular, fibrosis de órganos e inflamación. Se presenta más en mujeres en la quinta década y involucra factores genéticos y ambientales. Sus manifestaciones incluyen fenómeno de Raynaud, esclerodactilia, hipertensión pulmonar, úlceras digitales e involucramiento de pulmones, riñones e intestinos. El pronóstico depende del grado de afectación de órganos, siendo la enfermedad pulmonar inter