Descargar como PDF, PPTX



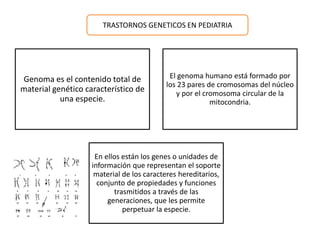
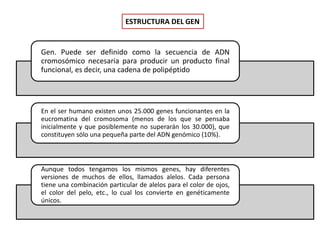

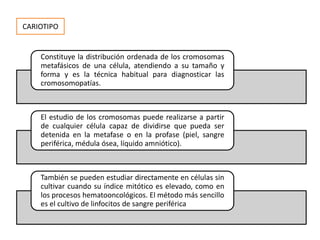
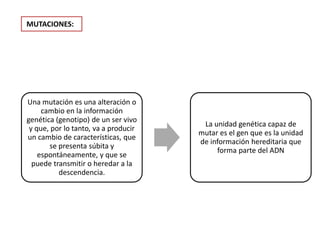
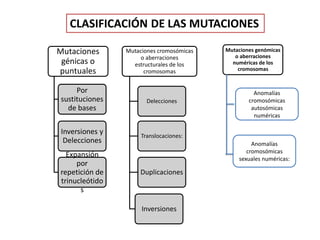




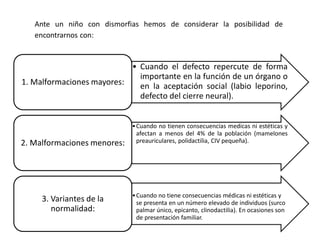



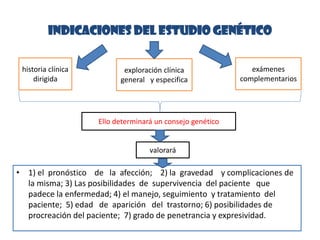








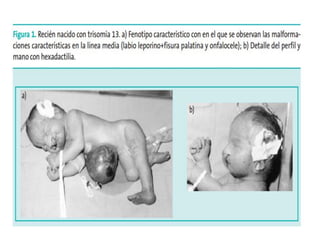



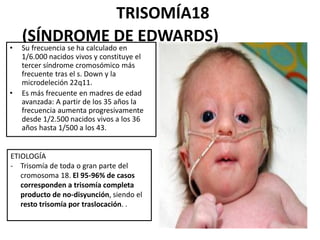
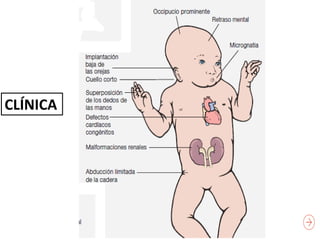






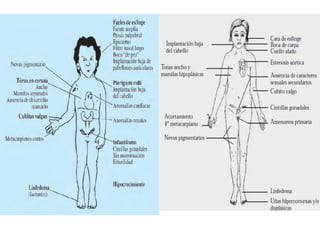
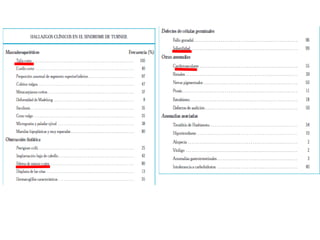


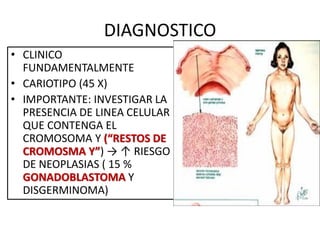



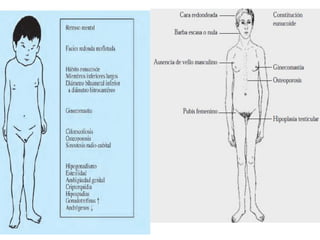
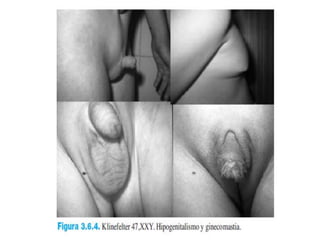
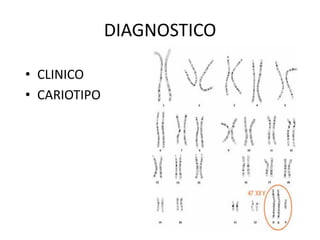


Este documento describe los aspectos fundamentales del estudio genético en pediatría. Explica que el estudio genético consiste en una historia clínica dirigida, exploración clínica y exámenes complementarios para evaluar rasgos dismórficos. El objetivo es establecer un diagnóstico y pronóstico, y proveer consejo genético sobre el riesgo de recurrencia y posibilidades de tratamiento y procreación. También describe las diferentes categorías de trastornos genéticos como las enfermedades monogénicas, c
















































































