Descargado 41 veces















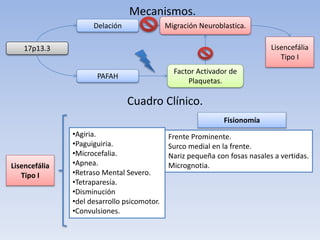



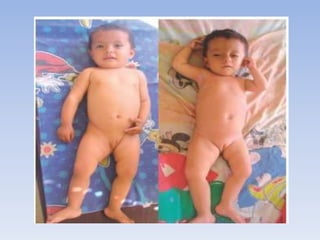












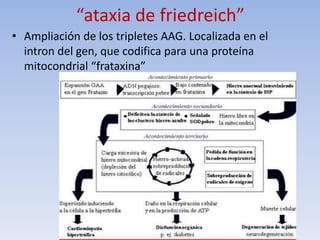




















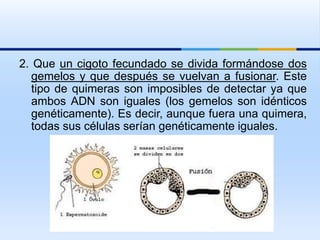
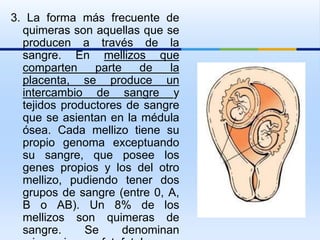






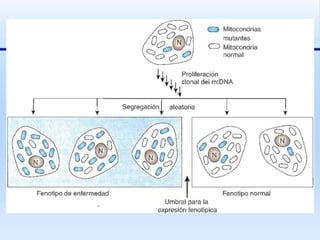








Este documento trata sobre diferentes síndromes y trastornos genéticos causados por mutaciones y rearreglos cromosómicos. En menos de 3 oraciones, resume que se describen las características clínicas y mecanismos moleculares de varios síndromes como el de Williams, Rubinstein-Taybi, Miller-Dreker y Russell-Silver, así como trastornos causados por expansiones de repeticiones como la enfermedad de Huntington y el síndrome del cromosoma X frágil. También se explican conceptos como la disomía un























































































