Descargado 119 veces


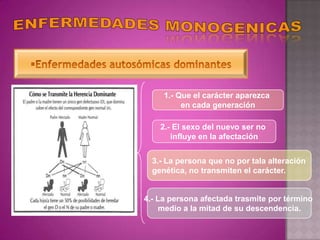
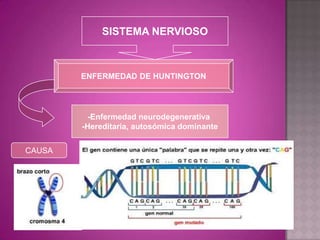





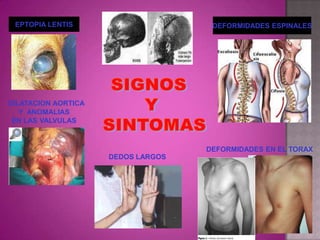
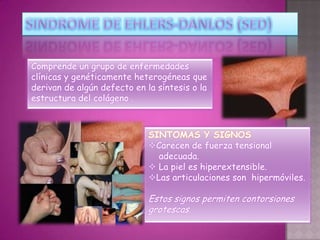

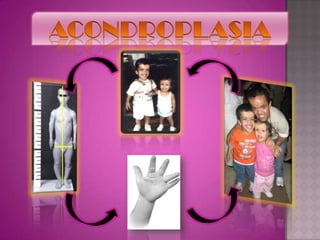

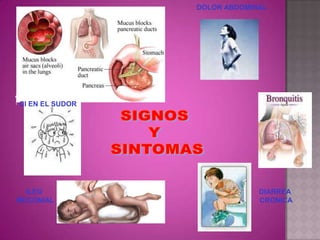




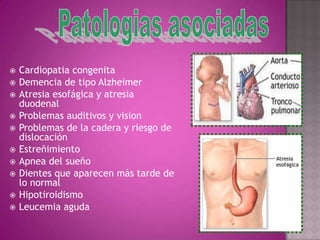

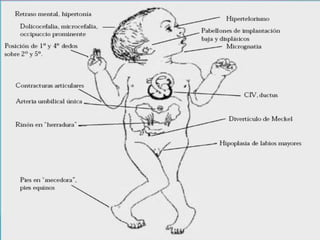
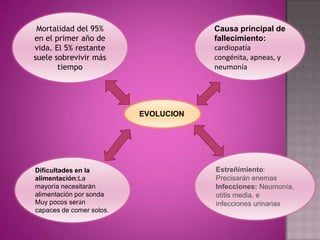

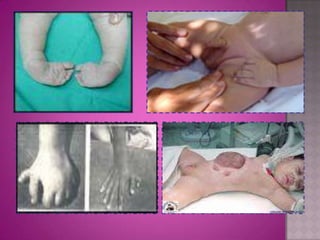
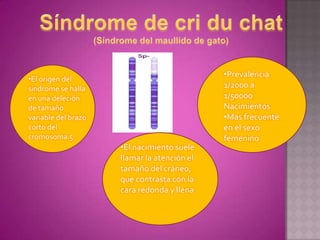

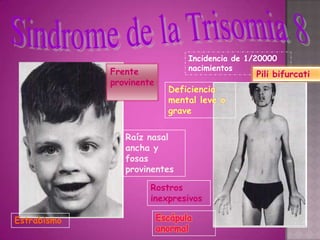

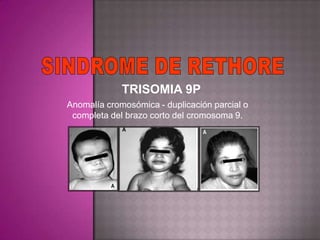

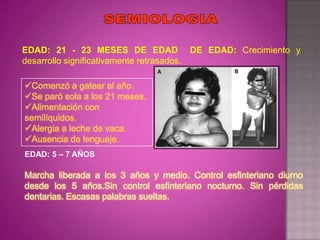


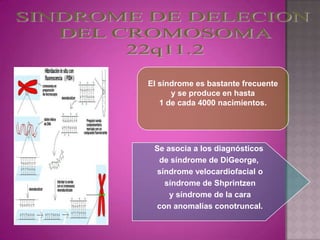
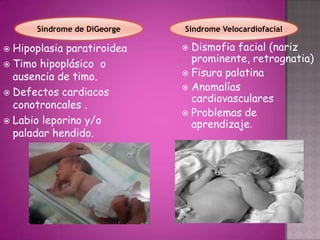


Este documento lista los integrantes de un grupo y describe varias enfermedades genéticas como la enfermedad de Huntington, fibrosis quística, enfermedad de Wilson y síndrome de Down. Las enfermedades discutidas son hereditarias y la mayoría son autosómicas dominantes o recesivas, lo que significa que los genes anormales se heredan de los padres. Se describen los síntomas y características de cada trastorno.






















































































![Resumen valoracion[1]2](https://cdn.slidesharecdn.com/ss_thumbnails/resumenvaloracion12-121116014413-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
