Descargado 172 veces






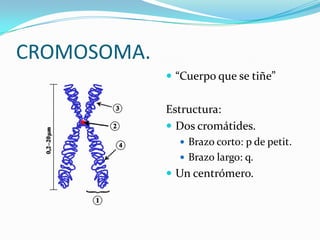















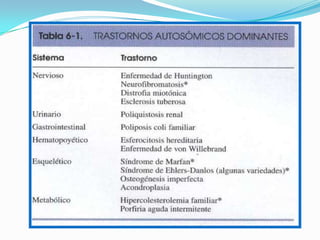



















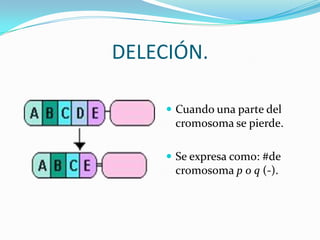
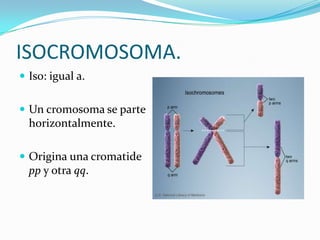





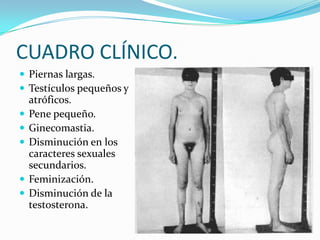



El documento aborda la estructura y función del ADN, genes, cromosomas y la herencia genética, explicando conceptos como mutaciones y trastornos mendelianos. Se detalla la diferencia entre trastornos hereditarios y congénitos, así como las características de diversas enfermedades genéticas y citogenéticas. También se menciona el síndrome de Down y Klinefelter, junto con sus etiologías, presentación clínica y diagnóstico.










































































































