Descargar para leer sin conexión



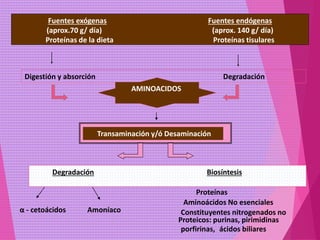
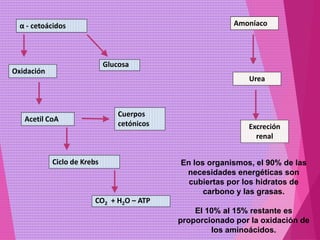
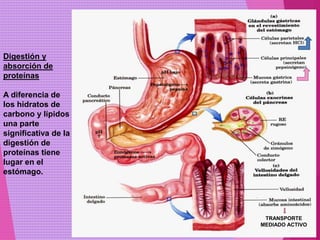
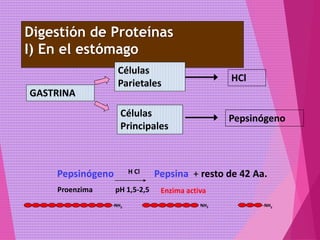

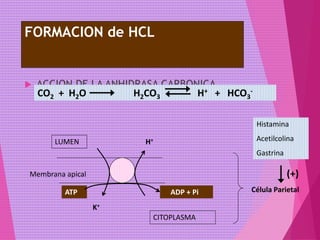
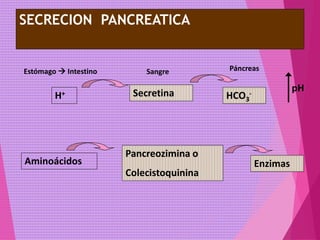
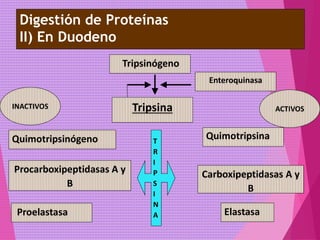









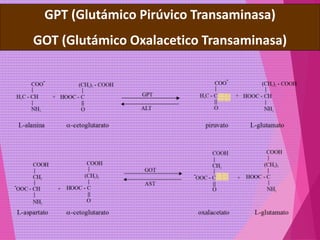
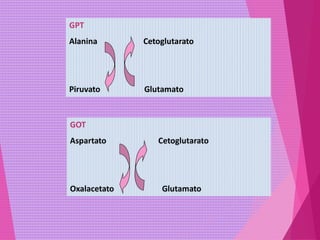
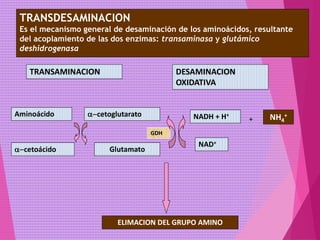





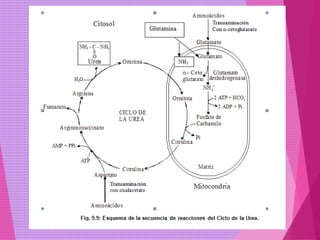




El documento trata sobre el metabolismo de las proteínas y los aminoácidos. Describe la digestión y absorción de proteínas, la biosíntesis y degradación de aminoácidos, y el metabolismo de los aminoácidos a nivel tisular. Explica los procesos de transaminación, desaminación y formación de urea para la eliminación de nitrógeno, así como las enfermedades relacionadas con trastornos en estos procesos.























































































