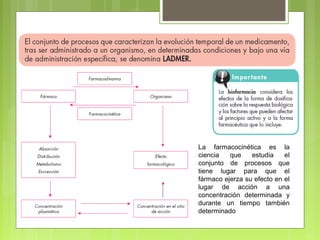
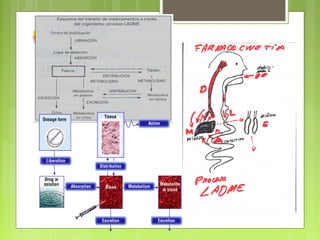
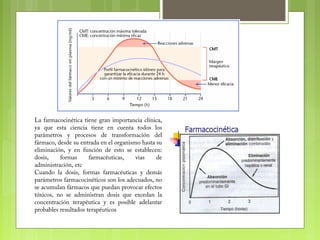
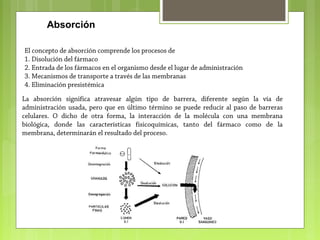
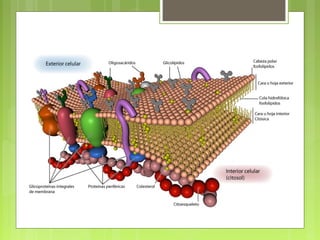
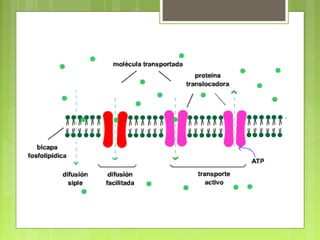
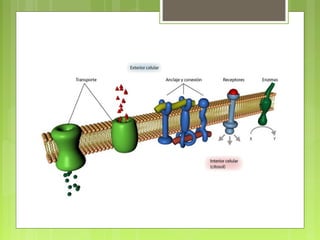
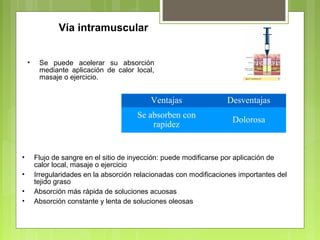
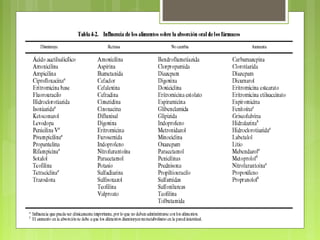
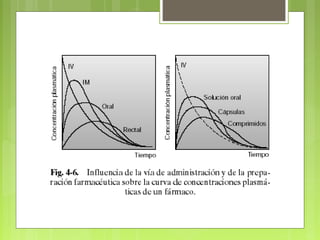
La farmacocinética estudia los procesos de absorción, distribución y eliminación de los fármacos en el organismo. Estos procesos determinan la concentración del fármaco en el lugar de acción y su efecto. La absorción implica la liberación del fármaco de su formulación farmacéutica y su paso a través de membranas biológicas mediante mecanismos como la difusión o el transporte activo.