








































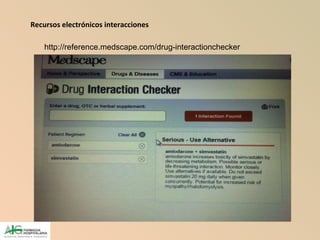







Este documento describe las interacciones farmacológicas entre medicamentos, las cuales pueden alterar su efectividad o causar efectos adversos. Explica que las interacciones ocurren cuando la presencia de un fármaco modifica la acción de otro, y analiza los mecanismos farmacodinámicos y farmacocinéticos involucrados. También destaca la importancia de estudiar las interacciones para evitar daños en pacientes polimedicados.











































































































