Descargado 119 veces













































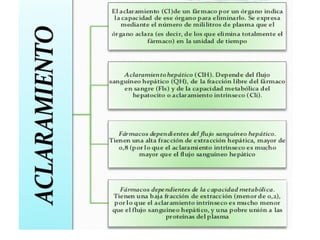






Este documento habla sobre los conceptos fundamentales de farmacocinética. Explica procesos como la absorción, distribución, metabolización y eliminación de los fármacos en el organismo. También describe factores que afectan estos procesos como la vía de administración, las características del fármaco, y condiciones fisiológicas del paciente. Finalmente, resalta la importancia de comprender la farmacocinética para optimizar el tratamiento farmacológico de cada individuo.











































































































