Descargar como PDF, PPTX





















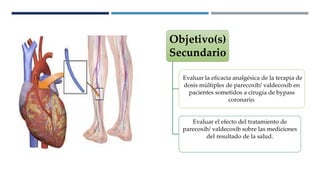




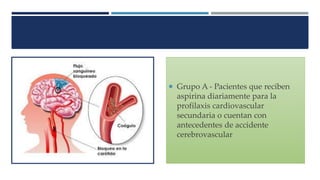

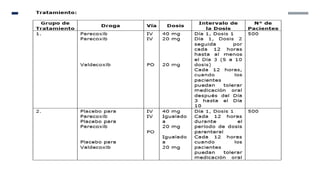





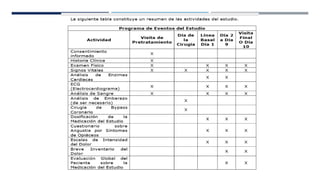









Este documento presenta el protocolo de un ensayo clínico multicéntrico, aleatorizado y doble ciego que evalúa la seguridad y eficacia analgésica de Parecoxib sódico seguido de Valdecoxib en comparación con placebo en pacientes sometidos a cirugía de bypass coronario. El objetivo primario es evaluar eventos adversos cardiovasculares y el objetivo secundario es evaluar la eficacia analgésica y efectos en resultados de salud. El estudio incluirá al menos 1000 pacientes estratificados en dos














































































