Descargado 313 veces













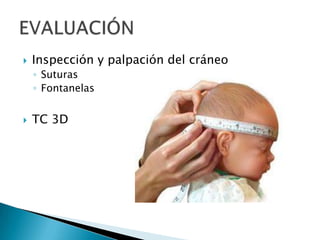






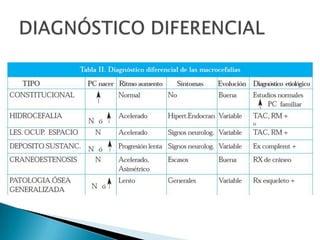


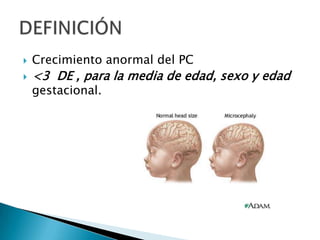







El documento describe las características anatómicas del cráneo al nacer, incluyendo fontanelas y suturas. Explica que la craneosinostosis es la fusión prematura de una o más suturas, pudiendo ser primaria o secundaria a otras condiciones. Detalla los tipos de craneosinostosis, síntomas, exámenes de diagnóstico y tratamiento quirúrgico temprano para evitar complicaciones. También aborda las causas y manifestaciones de macrocefalia y microcefalia, así como los exámen























































































