Descargado 42 veces










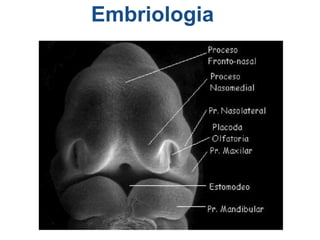
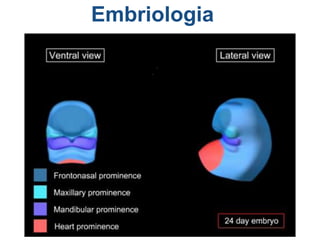




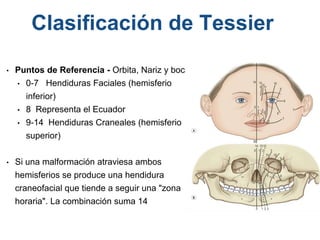
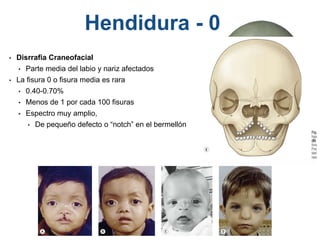
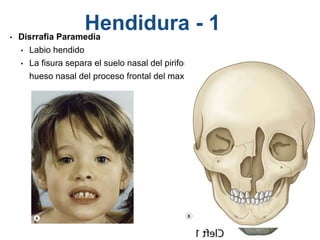
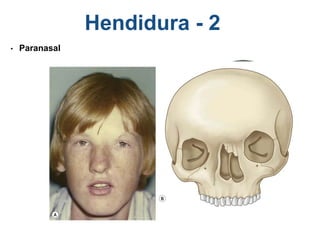
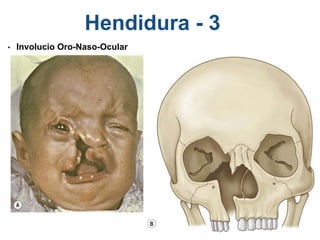
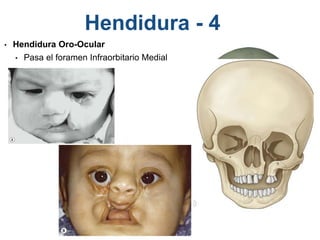
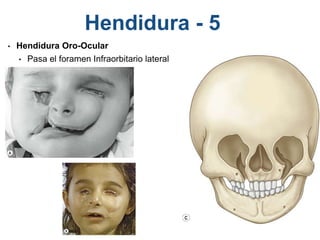
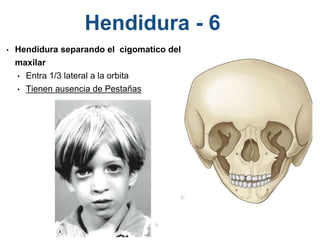
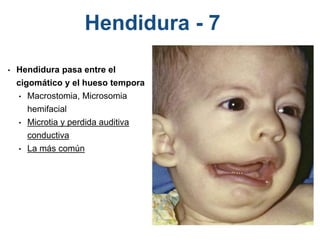
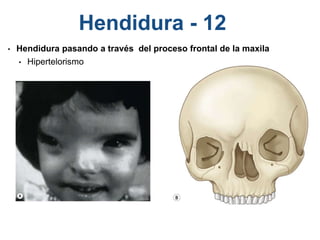
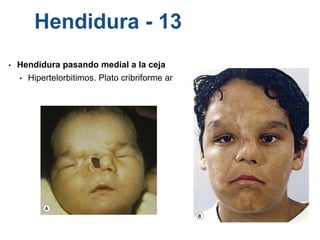
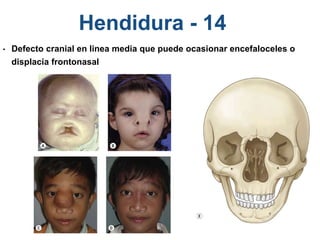
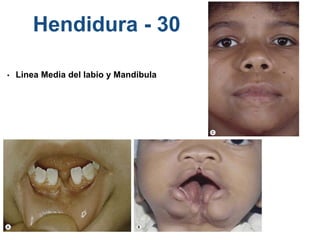
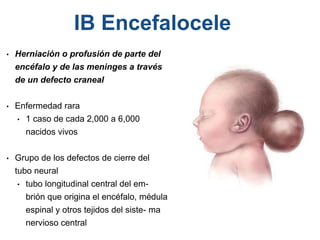
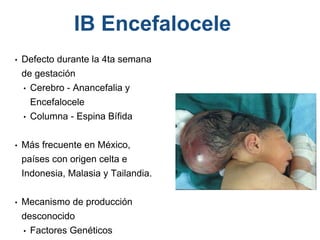
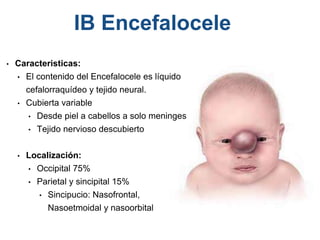

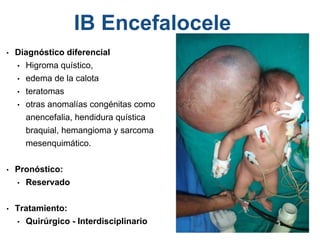
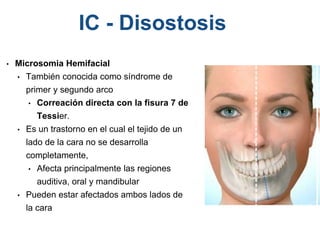

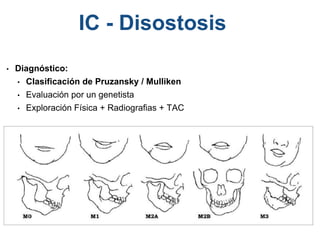
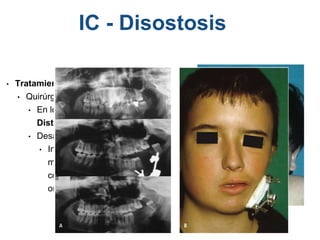
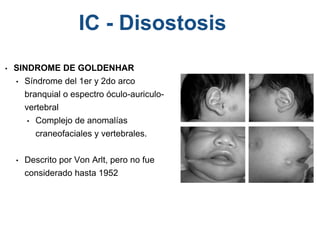



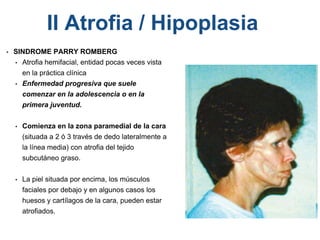
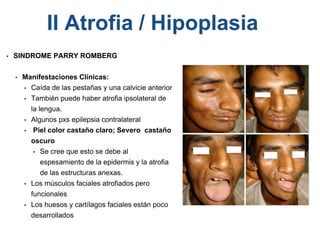

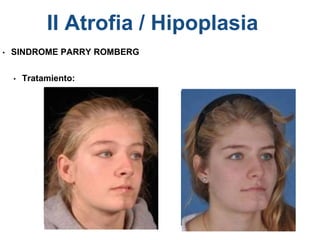
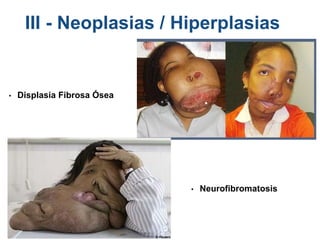

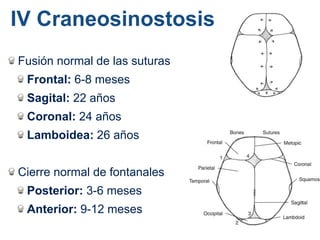


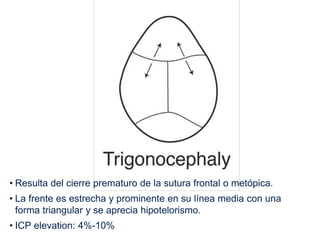
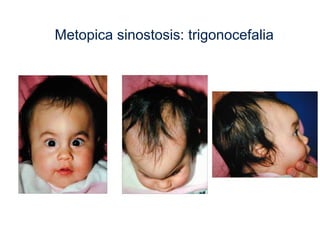
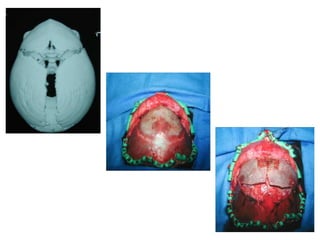
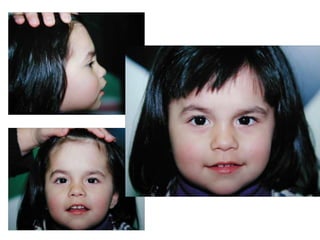
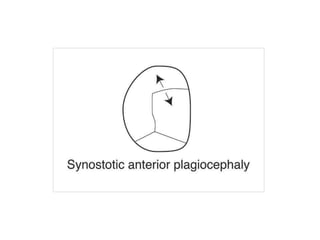
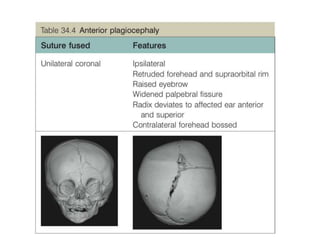
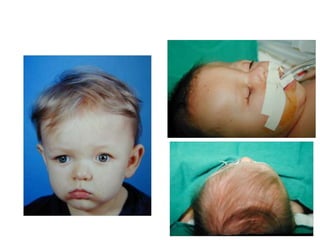
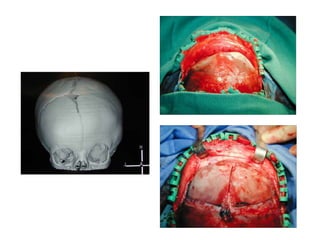
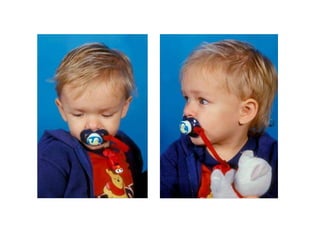
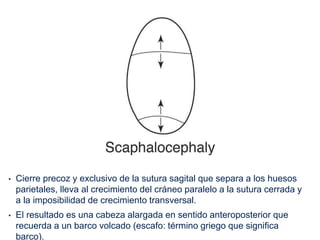
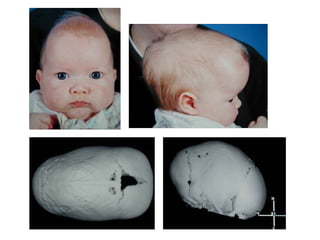
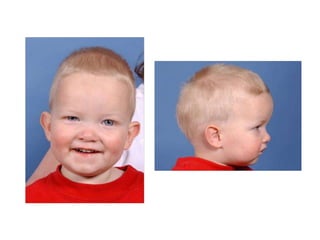
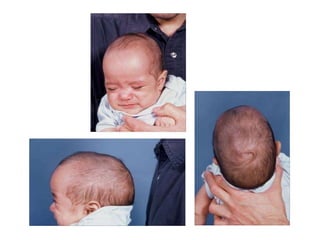
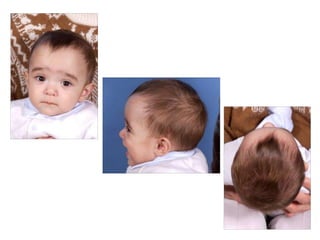


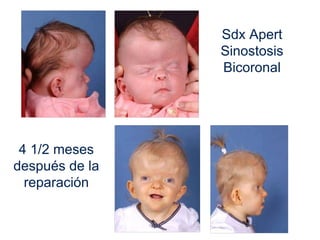

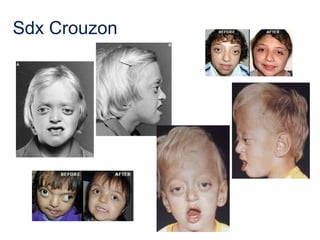




El documento detalla las malformaciones craneofaciales, incluyendo su frecuencia, clasificación y las implicaciones clínicas asociadas. Se discuten las causas, como factores genéticos y teratogénicos, y se presentan distintas clasificaciones como la de Tessier y Kárfík. Además, se abordan síndromes específicos como el de Treacher Collins y la microsomia hemifacial, enfatizando su diagnóstico y tratamiento quirúrgico multidisciplinario.















































