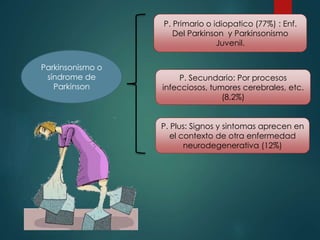
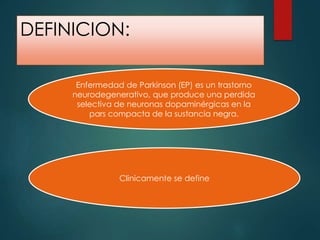
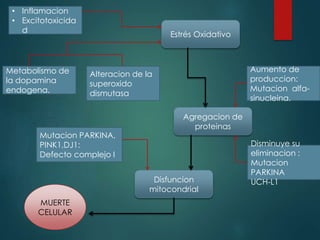
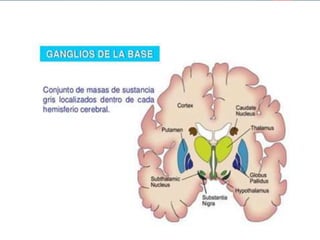
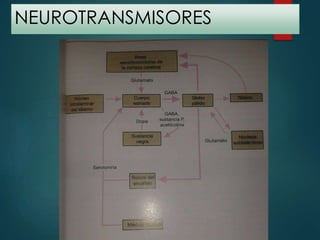
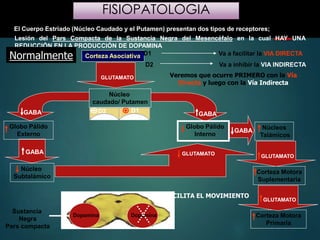
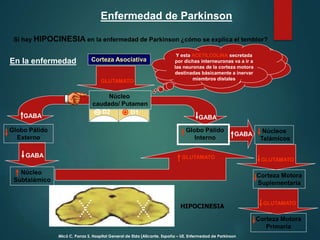
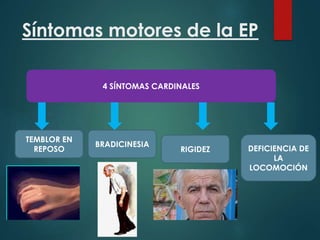
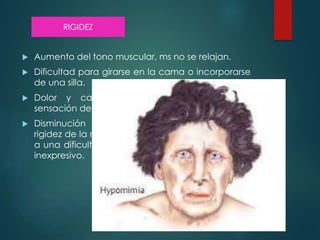
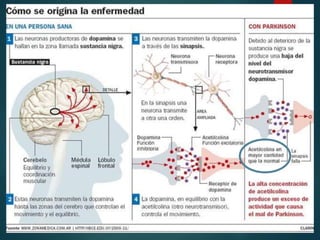
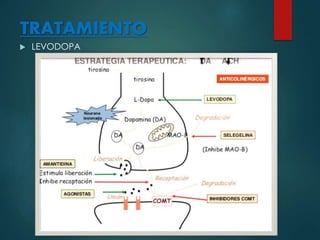
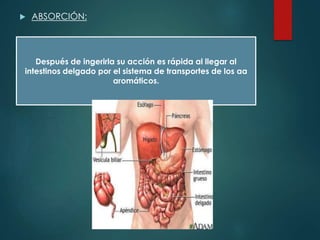
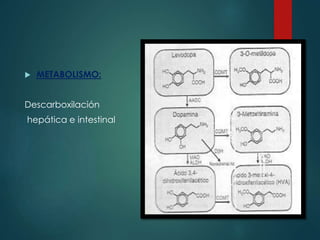
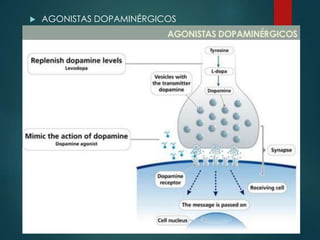
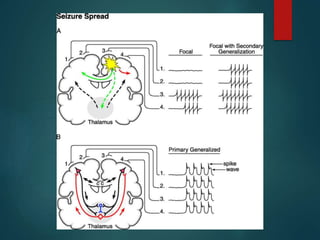
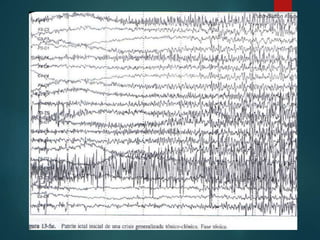
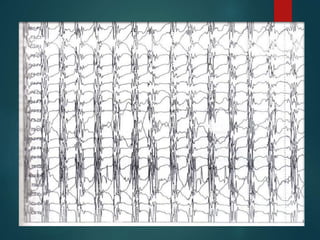
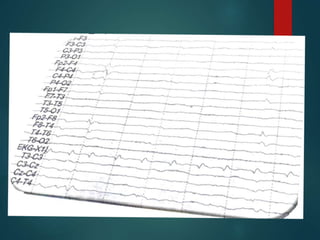
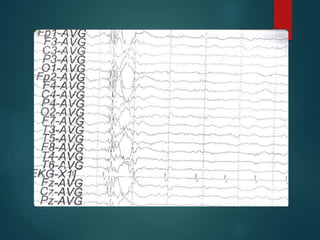
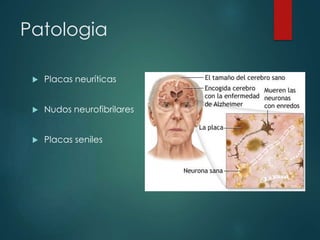
Este documento resume la enfermedad de Parkinson. En 3 oraciones: La enfermedad de Parkinson es un trastorno neurodegenerativo causado por la pérdida selectiva de neuronas dopaminérgicas en la sustancia negra. Sus síntomas principales son el temblor en reposo, la bradicinesia, la rigidez y la deficiencia en la locomoción. El tratamiento principal es la levodopa aunque también se usan agonistas dopaminérgicos.