Descargar para leer sin conexión



![EPILEPSY
An individual is considered to have epilepsy when
any of the following exists [5]:
●At least two unprovoked (or reflex) seizures
occurring more than 24 hours apart
●One unprovoked (or reflex) seizures and a
probability of further seizures similar to the general
recurrence risk after two unprovoked seizures (eg,
≥60 percent), occurring over the next 10 years. This
may be the case with remote structural lesions such
as stroke, central nervous system infection, or
certain types of traumatic brain injury.
●Diagnosis of an epilepsy syndrome](https://image.slidesharecdn.com/sindromeepilepticosdellactante-170818052242/85/Sindrome-epilepticos-del-lactante-3-320.jpg)



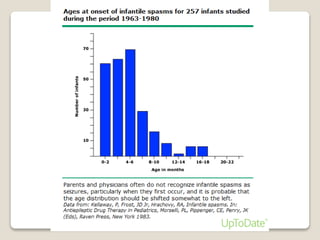































Este documento describe varios síndromes epilépticos que se presentan en lactantes. Describe el síndrome de West, caracterizado por espasmos epilépticos asociados con un patrón EEG de hipsarritmia. También describe el síndrome de Dravet, una epilepsia mioclónica grave de la infancia causada por una mutación genética. Otras condiciones discutidas incluyen el síndrome de Lennon-Gastaux, las crisis familiares benignas del lactante y la mioclónica benigna del lactante. Para cada una se



























![Estatus epileptico clase[1]](https://cdn.slidesharecdn.com/ss_thumbnails/estatusepilepticoclase1-131103081803-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)







































