1) El documento trata sobre sistemas de composición variable y equilibrio de fases. 2) Explica conceptos como sistema, propiedades, fase, equilibrio y presenta la relación fundamental entre las propiedades termodinámicas para mezclas homogéneas de composición variable. 3) El objetivo es determinar las propiedades termodinámicas para mezclas y resolver problemas de sistemas en equilibrio líquido/vapor considerando un comportamiento ideal.








![118
Punto de Ebullición Normal: Temperatura que produce en un
componente puro, una presión de vapor igual a una atmósfera.
Condiciones críticas: Condiciones a la cual las propiedades
intensivas de las fases líquido y vapor coexistentes, llegan a ser idénticas.
2.2. RELACIÓN FUNDAMENTAL ENTRE PROPIEDADES
TERMODINAMICAS PARA MEZCLAS HOMOGENEAS DE
COMPOSICIÓN VARIABLE:
La ecuación (2.1) relaciona la Energía Libre de Gibbs con la P y T de
cualquier sistema cerrado.
( ) ( ) ( )dT
nS
dP
nV
nG
d −
= (2.1)
Esta ecuación se aplica al caso de:
Sistema 1 sola fase.
Composición constante (No sufre reacción química)
Cerrado
Tenemos:
[∂ (nG)/∂P]T,n = (nV) y
[∂ (nG)/∂T]P,n = - nS
donde n indica que el número de moles de todos los componentes químicos
se mantienen constantes.](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-9-320.jpg)
![119
Para sistemas abiertos
1 sola fase.
Composición variable (Intercambios de masa con
los alrededores.
En este caso la energía libre de Gibbs no sólo es función de la P y T
sino también del número de moles (se toma o se agrega material al
sistema).
Entonces:
nG = g (P, T, n1, n2, ... , ni)
Donde ni: corresponde al número de moles de los componentes.
La diferencia total de (nG) es:
( ) ( )
[ ] ( )
[ ] ( )
[ ] i
n
,
T
,
P
i
n
,
P
n
,
T dn
n
nG
dT
T
nG
dP
P
nG
nG
d j
⋅
∂
∂
+
∂
∂
+
∂
∂
= ∑
Donde: la sumatoria ∑, se aplica a todos los componentes presentes.
nj: indica que se mantienen constantes todos los números de moles,
diferentes de ni.
Luego: Reemplazando las dos primeras derivadas por (nV) y (nS).
( ) ( ) ( ) ( )
[ ] i
nj
,
T
,
P
i dn
n
nG
dT
nS
dP
nV
nG
d ⋅
∂
∂
+
−
= ∑
(2.2)
2.3. POTENCIAL QUÍMICO DEL COMPONENTE I EN UNA MEZCLA (μi):
Se define como la variación de cualquier propiedad del sistema
con respecto al número de moles de un componente i en particular,](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-10-320.jpg)
![120
manteniendo la P, T y el resto de los componentes diferentes al componente
en particular (i) constantes.
( )
[ ] j
n
,
T
,
P
i
i n
nM ∂
∂
=
μ
M: representa cualquier propiedad del sistema. Para la Energía libre
de Gibbs (G): M = G
( )
[ ] j
P,T,n
i
n
nG
μi ∂
∂
= (2.3)
Luego la ecuación (2.2) se puede expresar en función del μi
( ) ( ) ( ) ∑μ
+
−
= i
dn
i
dT
nS
dP
nV
nG
d (2.4)
Esta ecuación representa la relación fundamental entre propiedades
para sistemas de una sola fase con masa constante o variable y composición
constante o variable.
2.3.1. EL POTENCIAL QUÍMICO COMO UN CRITERIO DE EQUILIBRIO:
Consideremos un sistema cerrado formado por 2 fases en equilibrio
(P, T son uniformes en todo el sistema), en el cual cada fase individual es un
sistema abierto (transferencia de masa de una fase a otra), Fig. 2.1](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-11-320.jpg)
![121
Fig. 2.1.- Sistema Cerrado en Equilibrio L-V.
Para cada fase en particular se escribe la Ecuación Energía Libre de
Gibbs para un sistema homogéneo de composición variable.
( ) ( ) ( ) ( )
[ ]
∑ ⋅
∂
∂
+
−
= i
n
,
T
,
P
i dn
n
nG
dT
nS
dP
nV
nG
d j
Fase α : d(nG)α
= (nV)α
dP – (nS)α
dT ∑ α
α
μ
+ i
i n
d
Fase α : d(nG)β
= (nV)β
dP – (nS)β
dT ∑ β
β
μ
+ i
n
d
i
Donde α y β identifican las fases.
El cambio total en la energía de Gibbs del sistema es la suma de las
ecuaciones anteriores. Toda propiedad total del sistema total se expresa con
una ecuación de la siguiente forma:
nM = (nM)α
+ (nM)β
Luego la Energía de Gibbs total del sistema.
d(nG) = d(nG)α
+ d(nVβ
)
( ) ( ) ( )
[ ] ( ) ( )
[ ] ∑ ∑ β
β
α
α
β
α
β
α
μ
+
μ
+
+
−
+
= i
i
i
i dn
dn
dT
nS
dT
nS
dP
nV
nV
nG
n
( ) ( ) ( ) ∑ ∑ β
β
α
α
μ
+
μ
+
−
= i
i
i
i dn
dn
dT
nS
dP
nV
nG
d](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-12-320.jpg)





![127
Esta ecuación representa el cambio de Entropía por mezclado para
gases ideales. Como 1/k > 1, el ΔS(gi)
siempre será > 0 (positivo). De
acuerdo a la Segunda Ley el proceso de mezclado para mezclas de gases
ideales a T, P constantes es irreversible y no se acompaña de transferencia
de calor.
Energía Libre de Gibbs: (Por definición)
G = H– TS
Para una mezcla de gas ideal se obtiene
Ggi
= Hg.i
– TSg.i
Si se sustituye en la ecuación (2.7) y (2.9).
∑ ∑ ∑
+
−
= ⋅
⋅
k
k
g
k
k
i
g
k
k
i
g
y
ln
y
RT
S
y
T
H
y
G
∑ ∑
+
= ⋅
⋅
k
k
i
g
k
k
i
g
y
ln
y
RT
G
y
G (2.10)
donde Gg.i
y son valores a T y P de la mezcla.
i
g
k
G ⋅
Potencial químico: ( µi)
El potencial químico del componente (i) de una mezcla de gas ideal se
expresa en función de la energía de Gibbs por la siguiente ecuación:
( )
[ ] j
n
,
T
,
P
i
i
g
i
g
i n
nG ∂
∂
=
μ ⋅
⋅
Multiplica la ecuación (2.10) por n:](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-18-320.jpg)
![128
∑ ∑
+
= k
k
i
.
g
k
k
i
.
g
y
ln
n
RT
G
n
nG
Como n
n
y k
k = ⇒ n
y
n k
k ⋅
= entonces ∑
= k
n
n
Tenemos
n
Ln
n
RT
y
Ln
n
RT
G
n
nG k
k
k
i
.
g
n
k
i
.
g
∑
∑
∑ −
+
=
∑ ∑ −
+
= n
Ln
RTn
n
Ln
n
RT
G
n
nG k
k
i
.
g
k
k
i
.
g
donde:
k: Es el conjunto de todos los componentes de la mezcla
i: Es un componente particular de la mezcla
Si del conjunto de todos los componentes de la mezcla se separa el
componente i a ese conjunto se le llama j.
o sea ∑ ∑ ∑
∑ −
=
⇒
+
= i
k
j
i
j
k n
n
n
n
n
n
luego
∑ ∑ −
+
+
+
= n
Ln
n
RT
n
Ln
n
RT
n
Ln
n
RT
G
n
G
n
nG j
j
i
i
i
.
g
j
j
i
.
g
i
i
i
.
g
Aplicando el concepto del y diferenciando a P y T constantes, los
nj son constantes.
i
.
g
i
μ
( )
[ ] ( ) ( )
[ ]
j
j
j n
i
n
i
i
n
i
i
i
i
.
g
i
i
.
g
i n
n
ln
n
n
ln
n
RT
n
ln
n
n
ln
n
RT
G ∂
∂
+
∂
∂
−
+
∂
∂
+
=
μ
Como: y
∑
= k
n
n ∑ +
= i
j n
n
n](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-19-320.jpg)







![136
Presión Total en Función de la Fase de Vapor:
( ) 1
sat
2
sat
1
sat
2 X
P
P
P
P −
+
=
Tenemos: ⇒
sat
1
1
1 P
X
P
Y = sat
1
1
1 P
P
Y
X =
( ) ( ) sat
1
1
sat
2
sat
1
sat
2 P
P
Y
P
P
P
P ⋅
−
+
=
( )
( ) sat
1
1
sat
2
sat
1
sat
2
sat
1 P
P
Y
P
P
P
P
P −
+
=
( ) P
Y
P
P
P
P
P
P 1
sat
2
sat
1
sat
2
sat
1
sat
1 −
+
=
( ) sat
2
sat
1
1
sat
2
sat
1
sat
1 P
P
P
Y
P
P
P
P =
−
−
( )
[ ] sat
2
sat
1
1
sat
1
sat
2
sat
1 P
P
Y
P
P
P
P =
−
+
( ) 1
sat
1
sat
2
sat
1
sat
2
sat
1
Y
P
P
P
P
P
P
−
+
= (2.26)
En la ecuación (2.26) se observa que P no varía linealmente con la
composición en la fase de vapor.
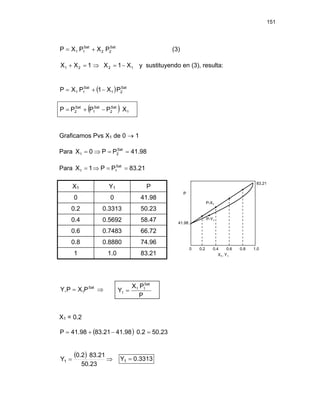
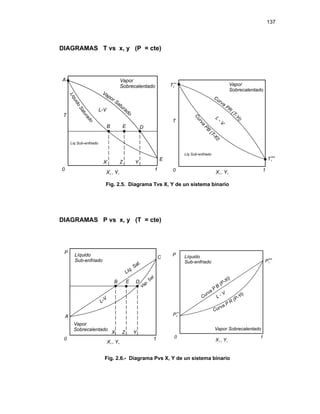
2.6.1. DIAGRAMAS DE FASES DE SISTEMAS BINARIOS:
El comportamiento de fases de sistemas binarios (i = 1,2) puede
representarse mediante diagramas Fig. 2.5 ; Fig. 2.6.
Estos diagramas generalmente se utilizan para ilustrar los estados de
mezclas binarias.
y
x,
vs
T y
x,
vs
P
Los diagramas: T vs x, y son a P = cte y
P vs x, y son a T = cte](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-27-320.jpg)








![147
− Balance de componente:
( ) V
Y
V
1
X
Z i
i
i +
−
= (i = 1, 2, … n) (2.35)
Sustituyendo Xi = Yi/ Ki en la ecuación (2.35)
Zi = Yi/Ki (1 – V) + Yi (V)
Zi = (Yi (1-V) + Yi V Ki)/Ki
Zi Ki = Yi (1 – V) + Yi V Ki
Zi Ki = Yi – Yi V + Yi V Ki ⇒ Zi Ki = Yi [1+V (Ki –1)]
Despejando Yi
( ) ( )
1
K
V
1
K
Z
Y i
i
i
i −
+
= (2.36)
Como la condición del Punto de Burbuja ∑ = 1
Yi
( ) ( )
∑ −
+ 1
K
V
1
K
Z i
i
i (2.37)
En la ecuación (2.37) se determina V por ensayo y error hasta que se
cumpla la condición.
Luego de (2.34) se obtiene L, y de (2.36) se calcula Yi
Los Ki se obtiene de la ecuación: P
P
K sat
i
i =
Luego como i
i
i X
Y
K = ⇒ i
i
i K
Y
X = (2.38)](https://image.slidesharecdn.com/tema2-220118160902/85/Tema-2-38-320.jpg)