
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Síndrome de Sjögren y otras patologías autoinmunitarias
Similar a Síndrome de Sjögren y otras patologías autoinmunitarias (20)
Último
Último (20)
Síndrome de Sjögren y otras patologías autoinmunitarias
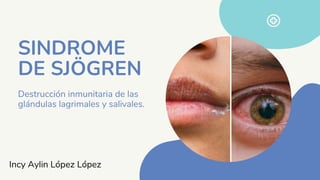
- 1. Destrucción inmunitaria de las glándulas lagrimales y salivales. SINDROME DE SJÖGREN Incy Aylin López López
- 2. Puede ser trastorno aislado (forma primaria) Asociado a enfermedades autoinmunitarias (forma secundaria)60%. También llamado síndrome seco: LES Polimiositis Esclerodermia Vasculitis, Enfermedad mixta del tejido conjuntivo Tiroiditis. Artritis reumatoide
- 3. PATOGENIA
- 4. Linfocitos T CD4+ colaboradores activados Linfocitos B Células plasmáticas. Infiltración linfocítica y de la fibrosis de las glándulas lagrimales y salivales= disminución de lagrimas y saliva. El resultado es la inflamación, el daño tisular y, finalmente, la fibrosis. 75% de los pacientes tienen factor reumatoide (anticuerpo reactivo frente a la IgG propia), coexista o no con la artritis reumatoide
- 5. Infiltración linfocítica periductal y perivascular Folículos linfoides con centros germinales. Hiperplasia de células epiteliales de conductos. Atrofia de los ácinos, fibrosis e hialinización. Sequedad del epitelio corneal, que se inflama, erosiona y ulcera Mucosa oral puede atrofiarse. con fisuras y úlceras inflamatorias Sequedad y las costras de la nariz pueden producir úlceras e incluso perforar el tabique nasal Morfología
- 6. Los ANA se detectan en el 50 al 80% de los pacientes. Han identificado una gran cantidad de anticuerpos específicos y no específicos de órgano. Más importante son: anticuerpos dirigidos contra dos antígenos de las ribonucleoproteínas, SS-A (Ro) y SS- B (La), que pueden detectarse hasta en el 90% de los pacientes mediante pruebas sensibles. Se consideran marcadores serológicos de la enfermedad. Porcentaje menor de pacientes con LES y, por ello, no son completamente específicos del síndrome de Sjögren.
- 8. Queratoconjuntivitis Mujeres de 50 y 60 años Xerostomía Manifestaciones extraglandulares: sinovitis, fibrosis pulmonar difusa y neuropatía periférica. Aumento de tamaño de la glándula parótida 60% se acompaña de otro trastorno inmunitario
- 9. Estadios precoces de la enfermedad: infiltrado inmunitario consta de una mezcla de linfocitos T y B policlonales. Reacción sin control: Tendencia de clones individuales dentro de la población de linfocitos B ventajas para crecer. Clon de linfocitos B dominante: Es habitualmente indicativa del desarrollo de un linfoma marginal, un tipo específico de neoplasia maligna de linfocitos B.
- 11. Se caracteriza por: 1) Inflamación crónica que se considera resultado de la autoinmunidad. 2) Daño generalizado de los vasos sanguíneos pequeños. 3) Fibrosis intersticial y perivascular progresiva de la piel y de múltiples órganos.
- 12. Esclerodermia difusa, caracterizada por una afectación extensa de la piel desde el comienzo, con progresión rápida y afectación visceral temprana. Esclerodermia limitada, afectación cutánea a menudo, en los dedos de las manos, los antebrazos y la cara, afectación visceral es tardía. Algunos pacientes sufren una combinación de calcinosis, fenómeno de Raynaud, alteración de la motilidad esofágica, esclerodactilia y telangiectasias, lo que se denomina síndrome CREST. 1 2
- 13. PATOGENIA
- 14. Linfocitos T CD4 que responden a un antígeno aún sin identificar se acumulan en la piel y liberan citocinas inflamatorias y fibroblastos, incluidos el TGF-P y la IL-13, pueden estimular la transcripción de genes que codifican el colágeno y/o tras proteínas de la matriz extracelular. Hay pruebas de activación inadecuada de la inmunidad humoral, y la presencia de varios autoanticuerpos, sobre todo de ANA que proporciona información diagnóstica y pronóstica. Autoinmunidad
- 15. ¿Acontecimiento iniciador o el resultado de la inflamación? Proliferación de la íntima en arterias digitales. Dilatación capilar con fuga, así como su destrucción. Asas capilares del lecho ungueal se distorsionan rápido en el curso de la enfermedad y después desaparecen. Liberación de mediadores que dañen el endotelio microvascular. Agregación plaquetaria= fibrosis Células musculares con mas receptores adrenérgicos. Lesión vascular
- 16. Fibrosis La fibrosis progresiva característica de la enfermedad. Culminación de múltiples anomalías, como la acumulación de macrófagos activados por la vía alternativa o tras la cicatrización de la lesión isquémica causada por las lesiones vasculares. Existen pruebas de que los fibroblastos de los pacientes con esclerosis sistémica presentan una anomalía intrínseca que les hace producir cantidades excesivas de colágeno.
- 18. Relación mujer:hombre de 3:1; incidencia máxima en grupo de edad de 50 a 60 años. Fenómeno de Raynaud en 70% de los pacientes Disfagia atribuible a la fibrosis esofágica y su hipomotilidad. Dificultades respiratorias por la fibrosis pulmonar= disfunción cardíaca derecha, y la fibrosis miocárdica
- 19. Proteinuria en hasta el 30% de los pacientes Más inquietante es la hipertensión maligna, con el consiguiente desarrollo de una insuficiencia renal Uno de ellos, dirigido contra la ADN topoisomerasa I es muy específico, el 10-20% con esclerosis sistémica difusa. Más posibilidades de tener una fibrosis pulmonar y una enfermedad vascular periférica. El otro, un anticuerpo anticentromérico, se encuentra en el 20-30% de los pacientes, que tienden a tener el síndrome CREST. Los pacientes con este síndrome tienen una afectación cutánea relativamente limitada, confinada a menudo a los dedos de las manos, los antebrazos y la cara Casi todos los pacientes tienen ANA: Se han descrito dos ANA
- 21. La enfermedad se caracteriza, desde el punto de vista serológico, por los títulos altos de anticuerpos frente a la ribonucleoproteína Ul. Debuta habitualmente con sinovitis de los dedos de las manos, fenómeno de Raynaud, miositis y afectación renal, que es moderada y responde bien a los corticoesteroides, al menos a corto plazo.
- 23. Caracterizadas por una inflamación necrosante de las paredes de los vasos sanguíneos que muestran indicios intensos de deberse a un mecanismo patogénico inmunitario. Vasculitis no infecciosas diferencia a estos trastornos de los debidos a la infección directa de la pared vascular sanguínea (como ocurre en la pared de un absceso)
- 25. Es una constelación de trastornos caracterizada por infiltrados tisulares dominados por células plasmáticas productoras de anticuerpos IgG4 y de linfocitos (sobre todo de linfocitos T), una fibrosis, una flebitis obliterante y, habitualmente, un aumento de la IgG4 sérica. La enfermedad afecta más a menudo a hombres de mediana edad y mayores. La patogenia de este trastorno no se conoce, aunque la producción de IgG4 en las lesiones es una característica fundamental de la enfermedad, no se conoce si este tipo de anticuerpo contribuye al trastorno.
- 27. Mecanismos de reconocimiento y rechazo de los aloinjertos El rechazo es un proceso en el que los linfocitos T y los anticuerpos producidos contra antígenos del injerto reaccionan contra los injertos tisulares y los destruyen.
- 28. Reconocimiento de aloantígenos del injerto por los linfocitos T y B Las principales diferencias antigénicas entre un donante y un receptor que dan lugar al rechazo de los trasplantes son las diferencias en los alelos del HLA. Tras el trasplante, los linfocitos T del receptor reconocen los antígenos del HLA del donante en el injerto por dos vías. Los antígenos del injerto bien se presentan directamente a los linfocitos T del receptor por las APC, son seleccionados por las APC del receptor, procesados (como cualquier otro antígeno extraño) y presentados a los linfocitos T del receptor.
- 29. Estas son las denominadas vías : directa e indirecta de reconocimiento de aloantígenos. Ambas conducen a la activación de linfocitos T C8+, que evolucionan a CTL, y linfocitos T CD4+, a su vez convertidos en células efectoras productoras de citocinas, principalmente linfocitos Th l. La vía directa puede ser la más importante para el rechazo agudo mediado por CTL. La vía indirecta puede desempeñar en ocasiones una función más destacada en el rechazo crónico.
- 30. Patrones y mecanismos de rechazo del injerto El rechazo del injerto se clasifica en hiperagudo, agudo y crónico, en función de sus características clínicas y patológicas.
- 31. Hiperagudo Es mediado por anticuerpos preformados específicos contra los antígenos de las células epiteliales del injerto. Los anticuerpos se unen a antígenos en el endotelio vascular del injerto y activan el sistema del complemento, produciendo lesión endotelial, trombosis y necrosis isquémica del injerto.
- 32. Agudo El rechazo agudo es mediado por linfocitos T y anticuerpos que son activados por aloantígenos en el injerto. Días o semanas después del trasplante principal causa de fracaso temprano del injerto. Rechazo celular agudo, los CTL CD8+ pueden destruir directamente las células del injerto, o bien los linfocitos CD4+ pueden secretar citocinas e inducir inflamación, que daña al injerto. Rechazo agudo mediado por anticuerpos (vascular, o humoral) los anticuerpos se unen al endotelio vascular y activan el complemento a través de la vía clásica. La inflamación y el daño endotelial consiguientes causan fracaso del injerto.
- 34. Crónico El rechazo crónico es una forma poco activa de daño del injerto que se produce a lo largo de meses o años y que condiciona la pérdida de función del injerto. Se manifiesta con fibrosis intersticial y estrechamiento gradual de los vasos sanguíneos del injerto (arterioesclerosís del injerto) Los causantes son linfocitos T que reaccionan contra los aloantígenos del injerto y secretan citocinas, que, a su vez, estimulan la proliferación y la actividad de fibroblastos y células de músculo liso vascular en el injerto.
- 36. Formas de aumentar la supervivencia del injerto
- 37. Inmunodepresores Fármacos inmunodepresores que se utilizan en la actualidad son los corticoesteroides (que reducen la inflamación), el micofenolato mofetilo (que inhibe la proliferación del linfocito) y el tacrolimús (FK506) Predecesor de la ciclosporina, inhibidor de la fosfatasa calcineurina, necesaria para la activación del factor nuclear de los linfocitos T activados (NFAT). El NFAT estimula la transcripción de genes de citocinas, en particular del gen que codifica el factor de crecimiento IL-2. De este modo, el tacrolimús inhibe las respuestas del linfocito T.
- 38. Otras estrategias Anticuerpos que eliminan a los linfocitos T y B, y las mezclas de IgG intravenosas (IGIV), que suprimen la inflamación por mecanismos desconocidos. Más reciente: Reducir respuestas inmunitarias contra el injerto para evitar que los linfocitos T del huésped reciban de las DC durante la fase inicial de la sensibilización. Se interrumpe la interacción entre las moléculas B7 situadas en las DC del injerto con los receptores CD28 de los linfocitos T del huésped administrando proteínas que se unan a los coestimuladores B7.
- 40. Las HSC se obtenían de la médula ósea, pero ahora se obtienen, habitualmente, de la sangre periférica después de movilizarlas de la médula ósea administrando factores de crecimiento hematopoyéticos, o de la sangre del cordón umbilical de niños recién nacidos, una fuente rica en HSC. Al receptor se le radia o trata con dosis elevadas de quimioterapia para destruir el sistema inmunitario.
- 42. Se produce cuando se trasplantan células inmunocompetentes o sus precursores a receptores con deficiencias inmunitarias, y las células transferidas reconocen aloantígenos en el huésped y atacan a sus tejidos.
- 43. EICH Aguda Las principales manifestaciones clínicas: se deben a la afectación del sistema inmunitario y de los epitelios de la piel, el hígado y el intestino. La afectación de la piel se manifiesta en forma de un exantema generalizado que puede llevar a la descamación en los casos graves. La destrucción de los conductos biliares pequeños da lugar a ictericia, y las úlceras mucosas en el intestino dan lugar a una diarrea sanguinolenta
- 44. EICH crónica También es frecuente la enfermedad hepática crónica que se manifiesta por ictericia colestásica. La lesión del tubo digestivo puede causar estenosis esofágicas. Sistema inmunitario está devastado, con una involución del timo y una pérdida de linfocitos en los ganglios linfáticos. Puede seguir al síndrome agudo u ocurrir lentamente. Lesión cutánea extensa, con destrucción de los anejos cutáneos y fibrosis de la dermis.
- 45. La inmunodeficiencia es una complicación frecuente del trasplante de HSC. La inmunodeficiencia puede ser el resultado del tratamiento previo, la mieloablación antes del trasplante de HSC, un retraso en la repoblación del sistema inmunitario del receptor y el ataque de los linfocitos injertados sobre las células inmunitarias del huésped. Inmunodeficiencia