
Recomendados
Más contenido relacionado
Similar a El Síndrome de Guillain-Barré.pdf
Similar a El Síndrome de Guillain-Barré.pdf (20)
Último
Último (20)
El Síndrome de Guillain-Barré.pdf
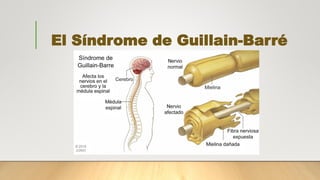
- 1. El Síndrome de Guillain-Barré
- 2. Alumna : Marlene Avalos Suarez Profesora: Rebeca Quintanilla Carrera: Enfermería Técnica IA
- 3. ¿Qué es el Síndrome de Guillain- Barré (SGB)? • es un trastorno poco frecuente en el cual el propio sistema inmunitario de una persona daña sus neuronas y causa debilidad muscular y a veces parálisis. El SGB puede causar síntomas que por lo general duran algunas semanas. La mayoría de las personas se recuperan totalmente del SGB, pero algunas padecen daños del sistema nervioso a largo plazo.
- 4. Síntomas • Los primeros síntomas consisten en debilidad u hormigueo, que suelen empezar en las piernas y pueden extenderse a los brazos y la cara. • En algunos casos puede producir parálisis de las piernas, los brazos o los músculos faciales. En el 20% a 30% de los casos se ven afectados los músculos torácicos, con lo que se dificulta la respiración. • En los casos graves pueden verse afectadas el habla y la deglución. Estos casos se consideran potencialmente mortales y deben tratarse en unidades de cuidados intensivos. • La mayoría de los casos, incluso los más graves, se recuperan totalmente, aunque algunos siguen presentando debilidad. • Incluso en los entornos más favorables, del 3% a 5% de los pacientes con el síndrome mueren por complicaciones como la parálisis de los músculos respiratorios, septicemia, trombosis pulmonar o paro cardiaco.
- 5. ¿Qué causa el SGB? • Se desconoce la causa exacta del SGB; no obstante, alrededor de dos tercios de las personas que desarrollan síntomas de SGB lo hacen varios días o semanas después de haber tenido diarrea o enfermedades respiratorias. • Las personas también pueden desarrollar SGB después de haber tenido influenza u otras infecciones (como el citomegalovirus y el virus de Epstein Barr).
- 6. ¿Quiénes corren con mayores riesgos de desarrollar el SGB? • Cualquier persona puede desarrollar SGB. Sin embargo, es más común entre los adultos mayores. La incidencia de SGB aumenta con la edad y las personas mayores de 50 años corren el mayor riesgo para el desarrollo de SGB
- 7. ¿Qué provoca el síndrome de Guillain-Barré? • pueden quedar dañados dos elementos del nervio. El primero de ellos es la vaina de mielina, una sustancia aislante que recubre el nervio y que permite que los impulsos nerviosos viajen rápidamente a los músculos. El otro elemento que puede quedar dañado en el ataque autoinmune es el axón, la parte del nervio que envía los mensajes. En Europa y Norteamérica, el 90% de los casos corresponden a la forma que daña la mielina (AIDP -, polineuropatía desmielinizante inflamatoria aguda) siendo la forma que afecta al axón (neuropatía axonal motora aguda -AMAN) más común en Asia y América Latina (30-50% de los casos).
- 8. Se caracteriza por una parálisis motora ascendente o descendente bilateral con relativa simetría, hiporreflexia o arreflexia, toma bulbar, no ocurrencia de trastornos sensitivos objetivos, presencia de síntomas disautonómicos y la parálisis respiratoria es la complicación más grave.2,3 Este síndrome se describió inicialmente por Landry en 1859 y posteriormente en 916 Guillain, Barré y Strohl4 además de los hallazgos del examen neurológico, encontraron un marcado aumento de la albúmina en el líquido cefalorraquídeo (LCR) sin pleocitosis. En décadas recientes con el advenimiento de los estudios electrofisiológicos se demostraron las alteraciones eléctricas que caracterizan la enfermedad.
- 9. Anatomía patológica • han demostrado que las lesiones aparecen en el sistema nervioso periférico, a cualquier nivel altitudinal del raquis y pares craneales; los sitios de lesión más constantes son las raíces anteriores y posteriores e incluyen las fibras intraganglionares. Estas lesiones son de tipo inflamatorio con infiltrado linfocitario y de macrófagos en las vénulas endoneurales y epineurales del sistema nervioso periférico. • Tempranamente en la enfermedad ocurre retracción del nodo de Ranvier, que produce un espacio internodal amplio, signos de degeneración de la vaina de mielina con segmentación y fagocitosis, que comienza en la región nodal y se dirige al núcleo de la célula de Schwann, lo que origina desmielinización segmentaria.
- 10. Cuadro clínico • Se caracteriza por la aparición de una deficiencia motriz, que puede acompañarse o no de parestesias en las manos o pies y de dolores en las extremidades y a lo largo del raquis. El defecto motor es relativamente simétrico, se inicia con mayor frecuencia en las extremidades inferiores y suele alcanzar progresivamente las superiores, el tronco, los músculos de la deglución, fonación y respiración; otras veces comienza por las extremidades superiores y se extiende a otros segmentos por encima o por debajo, y en ocasiones menos frecuentes comienza por los músculos de la fonación, deglución, masticación y respiración y desciende hacia las extremidades superiores, tronco y extremidades inferiores. En un tercio de los pacientes el defecto motor queda limitado a las extremidades inferiores, con muy ligeras manifestaciones en las superiores.
- 11. Tratamiento: • Nutrición: Debe ser por vía enteral; en los casos en que exista disfagia se sugiere alimentar a través de sonda nasogástrica; si ocurre disfunción parcial del intestino, pasar la sonda al yeyuno y alimentar a débito continuo. En cualquiera de los casos se deben cubrir los requerimientos nutricionales del paciente Control de la frecuencia cardíaca: Los episodios de taquicardia y bradicardia son generalmente transitorios y no merecen terapéutica, sólo se deberá intervenir en casos excepcionales en que se comprometa el gasto cardíaco.
- 12. Evaluación clínica: -Aumento de la frecuencia respiratoria, respiración superficial, inefectividad de la tos, respiración paradójica (bamboleo abdominal), capacidad de deglución afectada y disminución de la auscultación de los sonidos respiratorios. Analgesia; Debe practicarse con fármacos de baja potencia que no comprometan el centro respiratorio, ni empeoren la debilidad muscular.
- 13. Diagnóstico • puede ser difícil diagnosticar el Síndrome de Guillain- Barré en sus primeras etapas, puesto que varios trastornos tienen síntomas similares, por lo que los médicos deben examinar e interrogar a los pacientes y sus familiares cuidadosamente antes de hacer el diagnóstico. • Se debe observar si los síntomas son simétricos, la velocidad con la que aparecen los síntomas (en otros trastornos, la debilidad muscular puede progresar a lo largo de meses en vez de días o semanas), los reflejos(especialmente el reflejo rotuliano) usualmente desaparecen