
Recomendados
Más contenido relacionado
Similar a 15. BIODISPONIBILIDAD Y BIOEQUIVALENCIA_PARTE 3.pdf
Similar a 15. BIODISPONIBILIDAD Y BIOEQUIVALENCIA_PARTE 3.pdf (20)
Último
Último (20)
15. BIODISPONIBILIDAD Y BIOEQUIVALENCIA_PARTE 3.pdf
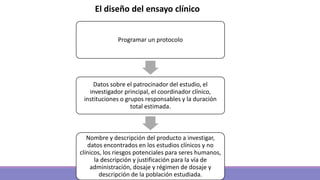
- 1. El diseño del ensayo clínico Programar un protocolo Datos sobre el patrocinador del estudio, el investigador principal, el coordinador clínico, instituciones o grupos responsables y la duración total estimada. Nombre y descripción del producto a investigar, datos encontrados en los estudios clínicos y no clínicos, los riesgos potenciales para seres humanos, la descripción y justificación para la vía de administración, dosaje y régimen de dosaje y descripción de la población estudiada.
- 3. Condición de administración de medicamentos Obtención y manipulación de las muestras biológicas -Por lo general la administración se realiza en ayunas -Verificar la ingesta de líquidos Con los tiempos de toma de muestra se decide tras una prueba piloto, en la que los tiempos seleccionados permitan delimitar todas las fases que configuran las curvas de niveles plasmáticos y éstos deberán seguirse hasta que haya pasado de 3 a 5 tiempos de vida
- 4. Número de voluntarios Criterios de inclusión y exclusión Es necesario conocer la variabilidad (coeficiente de variación) de los valores de AUC, esto basándose en un estudio piloto, de ensayo clínicos previos o datos publicados. Los voluntarios que sean sensibles a los fármacos de estudio deberán ser excluidos de igual manera a mujeres embarazadas o sujetos con enfermedades importantes porque no se los puede tener con tratamiento mientras se realiza el ensayo Los sujetos deben ser de ambos sexos, con una edad de 18 a 55 años de edad y peso que esté dentro del rango del 10-15% del peso ideal para su altura y sexo
- 6. •AUC, Cmax y Tmax Criterios de bioequivalencia Evaluación de enantiómeros
- 7. La seguridad y tolerancia Para la valoración de la tolerancia de un fármaco, se registran y describen temporalmente todos los efectos adversos comunicados espontáneamente por los voluntarios. Se recomienda que al inicio de cada visita y coincidiendo con la administración del fármaco y con cada una de las extracciones de sangre se pregunte sobre la posible aparición de efectos adversos. Valorar la causalidad de un evento adverso La valoración de la intensidad de la reacción adversa se hace también según una escala arbitraria, definida previamente de tres grados (leve, moderada, grave).
- 8. Consideraciones éticas Naturaleza y propósito del estudio Procedimientos a ser realizados Responsabilidades del voluntario como gozar de buena salud y no estar en tratamiento médico en el momento del estudio, acudir a las internaciones en las fechas y horarios informados Posibles riesgos y disconformidades como consecuencia de la administración de los fármacos Beneficios y compensaciones Indemnización, que deberá ser de acuerdo al tiempo dedicado en la realización del citado estudio clínico
- 9. ¿Por qué se pueden obtener diferencias en los ensayos de las formulaciones? 1. La secuencia u orden de administración de las formulaciones (efecto secuencia) 2. El período de administración (efecto período) 3. La formulación que se ha administrado (efecto formulación) 4. El posible efecto del fármaco administrado en el primer periodo sobre el fármaco administrado en el segundo período (efecto carry-over) 5. El diferente comportamiento del fármaco en los individuos (variabilidad interindividual) 6. Otros factores externos (variabilidad intraindividual)
- 11. ENSAYO IN VITRO: Estudio de disolución La disolución es una práctica que consiste en dispersar una sustancia en el seno de un líquido hasta nivel molecular o iónico. Los estudios de disolución se utilizan como herramientas para garantizar la calidad del producto, la uniformidad de diferentes lotes de producción, para determinar la bioequivalencia entre medicamentos genéricos y como guía para el desarrollo de nuevas formulaciones durante el proceso de fabricación Es una prueba que estima la liberación del principio activo a partir de la forma dosificada, evalúa la variabilidad interlote en cuanto a características de liberación y predice la biodisponibilidad y bioequivalencia de productos fabricados.
- 12. Pruebas de disolución La prueba consiste en determinar la cantidad de principio activo que se disuelve, luego de evaluarse un mínimo de 12 dosificaciones a disolución. Se debe determinar los perfiles de disolución en puntos múltiples de 2, 5, 10, 15, 20, 25, 30 minutos a diferentes velocidades de agitación, tipos de medios de disolución y temperaturas. Toma de muestra Comparar con el estándar
- 13. Velocidad de disolución Cantidad de fármaco que se disuelve en una unidad de tiempo a partir a partir de la forma farmacéutica sólida. 50, 75 y 100 rpm Controlada por un disolutor