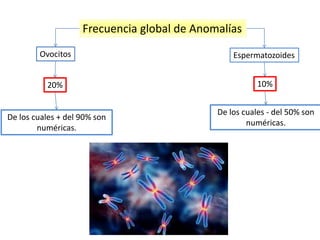
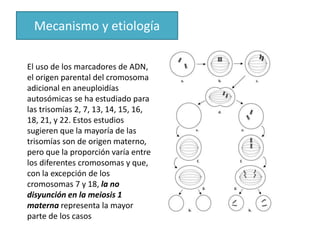
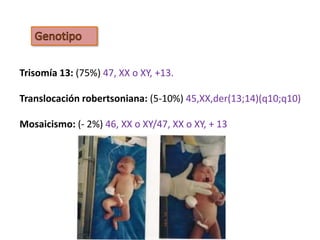
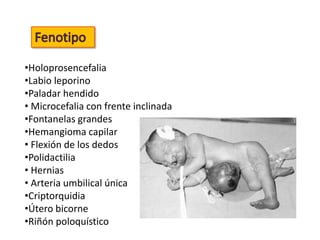
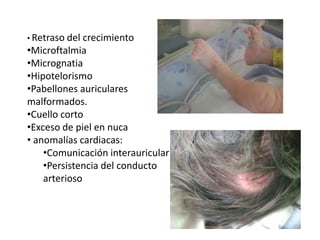
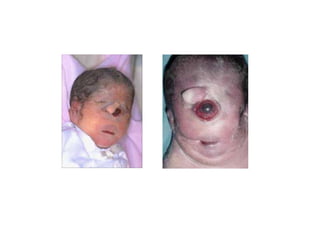
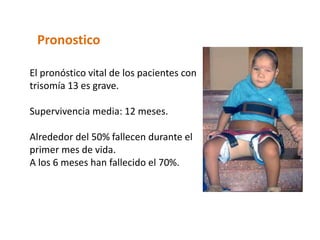
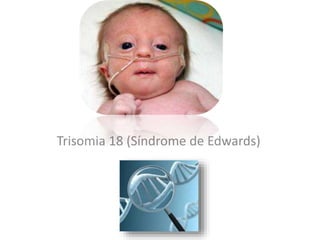
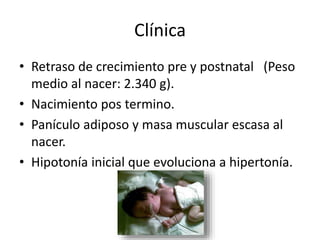
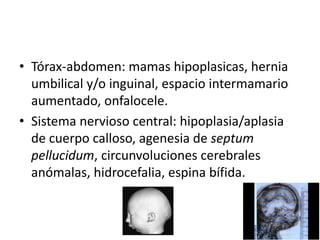
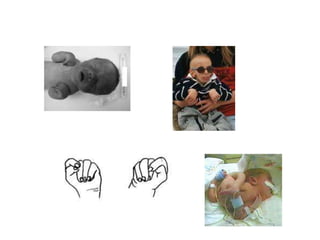
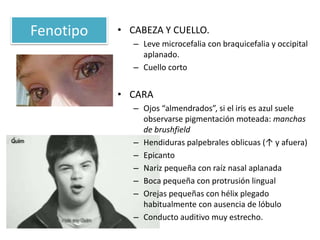
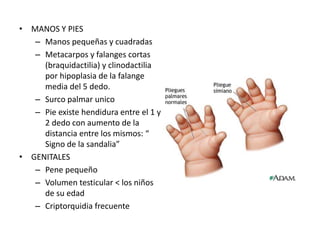
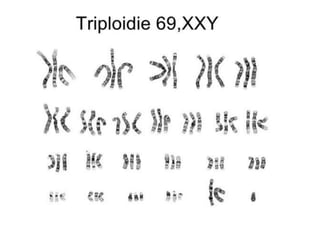
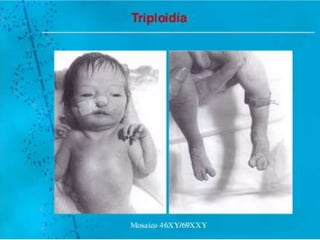
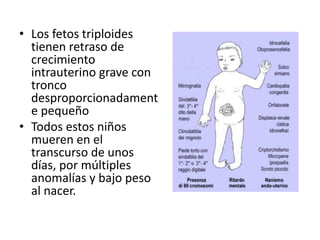
El documento describe las principales aneuploidías autosómicas como la trisomía 13, 18 y 21. Explica que la frecuencia global de anomalías cromosómicas es del 20% en ovocitos y 10% en espermatozoides, siendo la mayoría anomalías numéricas. Además, estudios con FISH han demostrado una frecuencia relativamente uniforme de aneuploidía autosómica en el esperma humano, aunque existe una mayor frecuencia de aneuploidía del cromosoma 21. Finalmente, señala que la mayoría de las tr