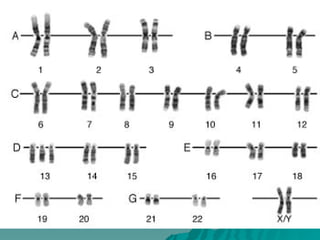
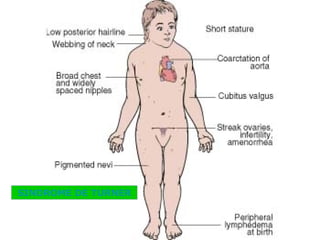
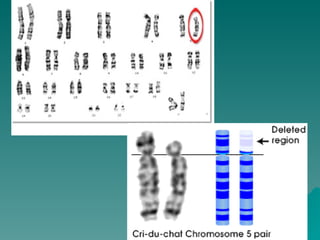
El documento presenta un resumen de los conceptos básicos de genética. Explica las leyes de Mendel sobre la herencia de caracteres dominantes y recesivos observadas en experimentos de cruzamiento de guisantes. También describe la estructura del ADN y cromosomas, así como diferentes síndromes genéticos causados por mutaciones y anomalías cromosómicas.