
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Examenes de laboratorio , enfermedades Hematologicas y Anestesia
Similar a Examenes de laboratorio , enfermedades Hematologicas y Anestesia (20)
Más de CLAUDIA VERONICA BOJORGE
Más de CLAUDIA VERONICA BOJORGE (20)
Último
Último (20)
Examenes de laboratorio , enfermedades Hematologicas y Anestesia
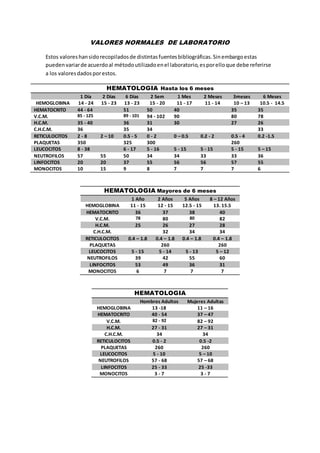
- 1. VALORES NORMALES DE LABORATORIO Estos valoreshansidorecopiladosde distintasfuentesbibliográficas.Sinembargoestas puedenvariarde acuerdoal métodoutilizadoenel laboratorio,esporelloque debe referirse a los valoresdadosporestos. HEMATOLOGIA Hasta los 6 meses 1 Día 2 Días 6 Días 2 Sem 1 Mes 2 Meses 3meses 6 Meses HEMOGLOBINA 14 - 24 15 - 23 13 - 23 15 - 20 11 - 17 11 - 14 10 – 13 10.5 - 14.5 HEMATOCRITO 44 - 64 51 50 40 35 35 V.C.M. 85 - 125 89 - 101 94 - 102 90 80 78 H.C.M. 35 - 40 36 31 30 27 26 C.H.C.M. 36 35 34 33 RETICULOCITOS 2 - 8 2 – 10 0.5 - 5 0 - 2 0 – 0.5 0.2 - 2 0.5 - 4 0.2 -1.5 PLAQUETAS 350 325 300 260 LEUCOCITOS 8 - 38 6 - 17 5 - 16 5 - 15 5 - 15 5 - 15 5 – 15 NEUTROFILOS 57 55 50 34 34 33 33 36 LINFOCITOS 20 20 37 55 56 56 57 55 MONOCITOS 10 15 9 8 7 7 7 6 HEMATOLOGIA Mayores de 6 meses 1 Año 2 Años 5 Años 8 – 12 Años HEMOGLOBINA 11 - 15 12 - 15 12.5 - 15 13. 15.5 HEMATOCRITO 36 37 38 40 V.C.M. 78 80 80 82 H.C.M. 25 26 27 28 C.H.C.M. 32 34 34 RETICULOCITOS 0.4 – 1.8 0.4 – 1.8 0.4 – 1.8 0.4 – 1.8 PLAQUETAS 260 260 LEUCOCITOS 5 - 15 5 - 14 5 - 13 5 – 12 NEUTROFILOS 39 42 55 60 LINFOCITOS 53 49 36 31 MONOCITOS 6 7 7 7 HEMATOLOGIA Hombres Adultos Mujeres Adultas HEMOGLOBINA 13 -18 11 – 16 HEMATOCRITO 40 - 54 37 – 47 V.C.M. 82 - 92 82 – 92 H.C.M. 27 - 31 27 – 31 C.H.C.M. 34 34 RETICULOCITOS 0.5 - 2 0.5 -2 PLAQUETAS 260 260 LEUCOCITOS 5 - 10 5 – 10 NEUTROFILOS 57 - 68 57 – 68 LINFOCITOS 25 - 33 25 -33 MONOCITOS 3 - 7 3 - 7
- 2. HEMATOLOGIA EN PEDIATRIA La sangre estáformada por dos fases: 1. Elementosformes 2. Plasma Los globulosoelementosformes de lasangre sonlos eritrocitosohematíes,lasplaquetasy losleucocitos. Los Eritrocitosohematíes suconcentraciónnormal esde 4.5 – 5.5 millones/mm3. Las plaquetas sonanucleadasyestánformadas de fragmentosde citoplasmade células gigantesde lamédulaósea,los megacariocitos. LEUCOCITOS RECUENTO LEUCOCITARIO Normalmente de 4.000 a 11.000 /mm3. Adultos/niños>2años:4.000 – 11.000/mm3. Niñosmenoresde 2años:6.200 – 17.000/mm3 ReciénNacidos:9.000 – 30.000/mm3 RN: 9.000 - 30.000/ mm3 1 – 3 años: 5.500 - 18.000/ mm3 4 – 7 años: 5.500 - 15.000/ mm3 8 – 13 años:4.500 - 13.500/ mm3 14 – 16 años:4.000 - 10.000/ mm3 Los leucocitosse clasificanengranulocitosópolimorfonucleares(PMN)ya granulocitos. Los granulocitosopolimorfonucleares(PMN)de acuerdoala afinidadtintorial de susgránulos citoplasmáticosse distinguen3tipos: 1. Neutrófilos 2. Eosinofilos 3. Basófilos Los agranulocitos (Notienen gránulos citoplasmáticos) existen2tipos: 1. Linfocitos 2. Monocitos
- 3. FORMULA LEUCOCITARIA NEUTROFILOS ABASTONADOS SEGMENTADOS PORCENTAJE RELATIVO 1 – 4% 40 – 60 % VOLUMEN ABSOLUTO 150 – 400 3.000 – 5.000 EOSINOFILOS 1 – 4% 20 – 350 BASOFILOS 0 – 4% 10 - 60 MONOCITOS 4 -8 % 100 -500 LINFOCITOS 20 -40 % 1500 - 4000 DIFERENCIAL DE GLOBULOS BLANCOS LACTANTES NIÑOS NEUTROFILOS JUVENILES BANDA SEGMENTADOS 0% Hasta 8% 17 – 60% 0% Hasta 6% 25 – 60% EOSINOFILOS 1 – 5% 1 – 5 % BASOFILOS 1% 1% LINFOCITOS 20 – 60% 25 – 60% MONOCITOS 1 – 6 % 1 – 6% NEUTROFILOS: Los neutrofilos muy jóvenes o juveniles son los llamados metamielocitos se caracteriza por poseernúcleo con una escotadura profunda que indica el inicio del proceso de formación de lóbulos. Los neutrófilos en BANDA, EN BASTON O SIMPLEMENTE BASTONADO son aquellos que antesde alcanzar la formalobuladatípicade la célulamadura,el granulocitopasapor una fase intermedia en la cual el núcleo tiene la forma de un cayado. El neutrófilo, después de tener su núcleo segmentado, es una célula en estado final de diferenciación, realizando síntesis proteica muy limitada.(SEGMENTADOS) Funciones Los neutrófilosconstituyenlaprimeralíneade defensacelularcontralainvasiónde microorganismos,siendofagocitosactivosde partículasde pequeñasdimensiones. DESVIACIÓN A LA IZQUIERDA:Se refiere ala presenciaensangre periférica de formas inmadurasesdecirse refiere al aumentodel númerode abastonados y la aparición en sangre periféricade elementos más jóvenes. Se observa en infecciones graves, intoxicaciones, etc. Causas: Procesos infecciosos agudos
- 4. Leucemias DESVIACIÓN A LA DERECHA: Se refiere al aumento de los neutrófilos segmentados (anemia perniciosa, sprue, anemia megaloblástica). Causas: Deficiencia de Vitamina B12 ó ácido fólico NEUTROFILIA: Esel aumentoanormal del númerode neutrófilosenel sistemacirculatorio. Aparte de la inflamación, casi cualquier factor susceptible de causar el menor grado de destrucción tisular originará neutrofilia. Una hemorragia aguda, una intoxicación, cualquier intervención operatoria. EOSINOFILOS Funciones: 1. Fagocitar y destruir determinados complejos de antígenos con anticuerpos 2. Limitar y circunscribir el proceso inflamatorio. Dicho proceso sirve de defensa, pero cuando se exagera puede perjudicar al organismo. Parece que una de las funciones del esosinofilo es realizar este tipo de fagocitosis selectiva. Los eosinofilos están dotados de movimientos ameboides y, como ocurre con las células ameboides en general, son atraídos por ciertas sustancias (Quimiotaxis). De este modo se concentranenlas áreasinflamadasyfagocitan más lentamenteque losneutrófilos,ysupoder para destruir microorganismos es pequeño. En lospacientesconafeccionesalérgicasaumentael número(EOSINOFILIA) de eosinofilos en la sangre.Estosgranulocitossonatraídosa laszonasde inflamaciónalérgicas gracias a factores quimiotacticos producidos por los mastocitos y granulocitos basófilos. En estas zonas, los eosinófilosliberanarilsulfatasa B e histaminasa, probablemente contenídas en sus gránulos, que destruyendosde losmediadoresquímicosde laalergia,laSRS-A (slow reacting substance of anaphylaxis) y la histamina. Hay también eosinofilia en los pacientes con parasitosis, hecho de importancia para el diagnóstico.Haypruebas de que loseosinófilos participan en la defensa contra los parásitos, como, por ejemplo, el Schistosoma mansoni. Los corticosteroides (hormonas de la capa cortical de la suprarrenal) provocan un descenso inmediatoenlaconcentraciónde loseosinófilosde lasangre yenlaszonas de inflamación, sin que se altere la concentración de estas células en la médula ósea. EOSINOFILIA: Puede ser idiopática, parasitaria, alérgica, pulmonar, paraneoplásica, endocrina, tóxica, en dermopatías, etc.
- 5. BASOFILOS La membranaplasmáticade losbasófilos,comolade losmastocitos,tiene tambiénreceptores para la inmunoglobulina E (Ig E). Liberan sus gránulos y las sustancias activas en ellos contenidos(heparinae histamina) al mediointercelular por la acción de los mismos estímulos que producen la expulsión de los granulos de los mastocitos. Sin embargo, y a pesar de sus semejanzas, los basófilos y los mastocitos presentan algunas diferencias en su origen y morfología. BASOFILIA: Su presencia es de interés al comienzo de las leucemias mieloides crónicas. Los basófilos aumentan en muchos procesos como en el MIXEDEMA, en el SÍNDROME NEFRÓTICO, DIABETES, etc. LINFOCITOS Funciones: Existendiferentestiposde linfocitos,yesposibledistinguirloslinfocitosporla presencia en su membrana de receptores integrados por inmunoglobulinas, especificas para numerosos antígenos, pero diferentes de un linfocito a otro. Existe una diferencia clara entre dos tipos de linfocitos. La célula precursora del linfocito se originaenla médulaóseay,en las aves, se hace inmunocompetente en la bolsa de Fabricio y en el timo. La bolsade Fabricioesunamasa de tejido linfático localizada en la cloaca de las aves. En ella, las indiferenciadas transportadas desde la médula ósea por la sangre son inducidas a dar origen a linfocitos capaces de ser transformados en plasmocitos, los cuales producen anticuerpos(inmunoglobulinas). Son los llamados linfocitos B, por que dependen de la bolsa de Fabricio,y estánrelacionadascon la defensa humoral del organismo a través de la síntesis de anticuerpos por los plasmocitos. En el timo,las células indiferenciadas llegadas de la médula ósea son inducidas a generar los linfocitos relacionados con la defensa inmunitaria celular del organismo. Estos son los llamados linfocitos T, por ser dependientes del timo. Cuandoentranencontacto con antígeno,loslinfocitosBse dividenyse diferencian en células plasmáticas, que sintetizan y secretan anticuerpos a la sangre, linfa y líquido intercelular. Ahora bien, los linfocitos T son los mas numerosos en la sangre. Estos linfocitos son responsables de las respuestas inmunitarias de base celular que no dependen de los anticuerpos circulantes. Un ejemplo es el rechazo de injertos. En este tipo de respuesta inmunitaria, los linfocitos T producen sustancias que actúan sobre las células transplantadas que estánpróximas.Poreste motivo,larespuesta de base celularsolo tiene lugar cuando los
- 6. linfocitos están localmente presentes para elaborar los factores que van a atacar las células extrañas. Hay que subrayar que laactividadde loslinfocitosesmuycomplejayque una de las funciones de los linfocitos T es facilitar la producción de anticuerpos por los linfocitos B. Ante ciertos antígenos, estos últimos sólo responden con la cooperación de los linfocitos T. Algunos linfocitos no entran en actividad al establecer contacto con el antígeno, pasando a formar parte de las células de la memoria inmunológica. Gracias a estas células, cuando un antígenoinvade el organismoporsegundavez,larespuestainmunitaria suele ser mucho más intensa y rápida. LINFOCITOSIS: Se destacan de entre todas: a) Infecciosas agudas: Entre ellas se citará como patología especial la mononucleosis infecciosa, infección frecuente en niños y que se debe diferenciar de las leucemias agudas con blastos. b) Otras linfocitosis: Postinfecciosa, en las hemopatías, endocrina, tóxica, carencial, metabólica, por radiaciones, etc. MONOCITOS: Los monocitos de la sangre representan una fase en la maduración de la célula mononuclear fagocitaria originada en la médula ósea. Esta célula pasa a la sangre, donde permanece sólo unosdías y , atravesandolaparedde loscapilaresyvénulas,penetraen el tejido conjuntivo y enalgunosórganos,transformándose enmacrófago,loque constituye una fase más avanzada de la célula mononuclear fagocitaria. Así, el monocito forma parte del sistema mononuclear fagocitarioosistemahistocitario.Comoocurre contodoslosleucocitos,exceptoloslinfocitos, losmonocitosnovuelven a la sangre una vez que atraviesan la pared de los vasos y penetran en los tejidos. MONOCITOSIS: En afeccionesdel sistemaretículoendotelial (aparición de los macrófagos hísticos). Aparecen en procesos crónicos y en la fase de defensa de los agudos) ERITROCITOS Hombres : 5.0 ±0.8 millones/mm3 Mujeres: 4.8 ±0.6 millones/mm3 Niños : 4.1- 5.1 millones/mm3 Menor de 1 mes: 3.5- 5.9 millones/mm3 1 – 12 meses: 3.7- 5.3 millones/mm3 1 – 16 años: 3.9- 5.1 millones/mm3
- 7. Los eritrocitos o hematíes en el hombre tienen el núcleo tienen la forma de un disco bicóncavo, con 7.2 µm de diámetro y 2.1 µm de espesor junto a su borde. La forma bicóncava proporcionaunagran superficie,loque facilitaloscambios de gases, ya que ningún punto del eritrocito dista más de 0.85µm de su superficie. Los hematíes con más de 8µm y los que tienen menos de 6µm de diámetro se denominan macrocitosy microcitos, respectivamente. Cuando la sangre contiene un elevado porcentaje de eritrocitos con dimensiones anormales, se dice que existe aniso-citosis. El eritrocito es muy flexible, lo que le permite adaptarse a la forma a veces irregular de los capilares. RETICULOCITOS Los eritrocitosal entrarenla corriente sanguíneadesde lamédulaósearoja, donde se forman, los glóbulos rojos contienen aún cierta cantidad de ribosomas. Cuando se coloran, estos corpúsculos presentan una coloración azulada debida a la basofilia del RNA, que predomina sobre la acidofilia de la hemoglobina. Algunos colorantes, como el azul brillante cresil, precipitan este RNA, dando origen a una delicada red de material basófilo que aparece bien coloreada en azul. Estos corpúsculos se llaman RETICULOCITOS, y su concentración en la sangre normal es de cerca del 1% del número total de hematíes. Los RETICULOCITOS permanecer cerca de 24 horas en la sangre circulante, tiempo que precisan para transformarse en hematíes completamente maduros. El recuento de RETICULOCITOS ES DE 0.5 – 1.5% DEL NUMERO DE ERITROCITOS. Son Hematies jovenes, inmaduros,Tamaño superior a los macrocitos con Tincion especial vital con azul cresil:sustancia granulofilamentosa en forma de red citoplasmatica (restos arn ribosomal).Son teñidos con Tenida con metodo de giemsa:tono azulado tono azulado (macrocitos policromados). Normalmente 5-15/1000 hematies =25.000-50.000 aprox El reticulocitoesel mismoeritrocitopolicromatofilo visto después de una tinción común, son capaces de sintetizar proteínas a un nivel importante de modo que también deben contener ARNm y ARNt, ARNr. El recuento de reticulocitos se mantiene alto día tras día puede suponerse que ha aumentado el grado de destrucción de eritrocitos o que estos están saliendo de la circulación por alguna otra vía. En estados hiperregenerativos de la eritropoyesis. (Hemorragias y hemoliticas).Disminuye en estados arregenerativos: aplasias carenciales (ferropenicas o megaloblasticas El recuento de reticulocitos obtiene información sobre el nivel de producción de eritrocitos, glóbulos rojos y el nivel de su destrucción o pérdida.
- 8. RETICULOCITOSIS: Es una dato característico de anemia hemolítica, refleja la capacidad de respuesta de la médula ósea. 1. Si el hematocrito está aumentado y la reticulocitosis es sostenida. La reticulocitosis que persiste de 5% o más (Corregida) con hematocrito estable varias semanas = proceso hemolítico continuo. 2. Anemias Hemolíticas congénitas tienen riesgo de supresión transitoria del funcionamiento de la médula ósea por infección viral. 3. Pueden tener CRISIS APLASICA CON DESTRUCCION PRECIPITADA DEL HEMATOCRITO Y RECUENTO BAJO DE RETICULOCITOS. 4. RETICULOCITOSIS + HTO CADA VEZ MAYOR SUGIERE EPISODIO HEMOLÍTICO TRANSITORIO COMO EN LA DIFERENCIA DE 6GPD CON EXPOSICIÓN A UN FARMACO OXIDANTE O RECUPERACION LUEGO DE LA PÉRDIDA SANGUINEA. CUERPOS DE HOWELL JOLLY En algunas condiciones patológicas ocurre a veces que fragmentos nucleares (con DNA) permanecen en el eritrocito después de la expulsión de su núcleo. Estos restos nucleares son Feulgen positivos y se tiñen por los colorantes básicos. Muchas veces adoptan la forma de uno o dos gránulos pequeños ( 1µm de diámetro) y reciben el nombre de cuerpos de Howell-Jolly. Cuando aparecen como filamentos circulares se llaman anillos de Cabot Causas: 1. HIPOESPLENISMO 2. ESPRUE IDIOPATICO CON O SIN ANEMIA MEGALOBLASTICA. HEMOGLOBINA: La hemoglobina es una heteroproteín de la sangre de peso molecular de 68,000 de color rojo característico, que transporta el oxígeno desde los órganos respiratorios hasta los tejidos en mamíferos ovíparos y otros animales. La forman cuatro cadenas polipeptídicas a cada una de las cuales se un grupo hemo,cuyo átomo de hierro es capaz de unirse de forma reversible al oxígeno. El grupo hemo se forma por:
- 9. 1. Unión de la Succinil CoA (Formado en el ciclo de Krebs o ciclo del ácido crítico) a un aminoácido glicina formado un grupo pirrol. 2. Cuatro grupos pirrol se unen formando la protoporfirina IX. 3. La protoprofirina IX se une a una molécula de hirro ferroso(Fe2+) formando el grupo hemo. La molécula de hemoglobina (proteína conjugada) está formada por 4 grupos hemo unidos a la globina, proteína del grupo de las histonas. El grupo hemo es un derivado porfirínico, que contiene un radical de hierro. Debido a variaciones de las cadenas de globina, se distinguen varios tipos de hemoglobina, de las cuales tres son consideradas normales: las hemoglobinas A1, A2 y F. La hemoglobina A1 (Hb A1) representa el 97% y la hemglobina A2 (HbA2), el 2% de la hemoglobina del adulto normal. El tercer tipo de hemoglobina normal es característica del feto, siendo conocida como hemoglobina fetal o F (Hb F). Representa el 100% de la hemoglobina del recién nacido, y su nivel baja progresivamente hasta el octavo mes, cuando alcanza un 1% porcentaje semejante al del adulto. La hemoglobina fetal muestra gran avidez por el oxígeno y tiene un papel importante en la vida prenatal, donde hay escasez de éste, pues el feto no tiene acceso al aire y obtiene oxígeno de los eritrocitos de la sangre materna a través de la placenta. En los pulmones, donde la presión de oxígeno es alta, cada molécula de hemoglobina se combina con cuatro moléculas de hemoglobina se combina con cuatro moléculas de oxígeno (una molécula de oxígeno para cada Fe de la hemoglobina), formándose la oxihemoglobina. Esta combinación es reversible , y el oxígeno transportado por la hemoglobina es transferido a los tejidos, donde la presión de oxígeno es baja. La combinación de la hemoglobina con el CO2 normalmente producido en los tejidos también es reversible y constituye la carbamino-hemoglobina. Pero la mayor del CO2 es transportada de los tejidos a los pulmones, disuelta en el plasma. Cuando la hemoglobina está unida al oxígeno se denomina Oxihemoglobina o hemoglobina oxigenada, dando el aspecto rojo o escarlata intenso característico de la sangre arterial. Cuando pierde el oxígeno, se denomina hemoglobina reducida y presenta el color rojo oscuro o bordó de la sangre venosa (se manifiesta clínicamente por cianósis). TIPOS DE HEMOGLOBINA
- 10. 1. Hemoglobina A o Hb A es llamada también hemoglobina del adulto o hemoglobina normal representa aproximadamente el 97% de la hemoglobina degradadaen el adulto, formada pordos globinas alfa y dos globinas beta. 2. Hemoglobina A2 Representa menos del 2.5% de la hemoglobina después del nacimiento formada por dos globinas alfa y dos globinas delta, que aumenta de forma importante en la beta-talasemia, alno poder sintetizar globinas beta. 3. Hemoglobina S: Hemoglobina alterada genéticamente presente en la Anemia de células falciformes. Afecta predominantemente la población aformericana y amerindia. 4. Hemoglobina t 5. Hemoglobina f: Hemoglobina característica del feto. 6. Oxihemoglobina: Representa la hemoglobina que se encuentra unida al oxígeno normalmente (Hb+O2) 7. Metahemoglobina: Hemoglobina con grupo hemo con hierro en estado férrico, Fe (III) (es decir, oxidado). Este tipo de hemoglobina no se une al oxígeno. Se produce por una enfermedad congénita en la cual hay deficiencia de metahemoglobina reductasa, la cual mantiene el hierro como Fe (III). La metahemoglobina tambien se puede producir por intoxicación de nitritos, por que son agentes metahemoglobinizantes. 8. Carbaminohemoglobina: Se refiere a la hemoglobina unida al CO2 después delintercambio gaseoso entre los glóbulos rojos y los tejidos (Hb+CO2). 9. Carboxihemoglobina: Hemoglobina resultante de la unión con el CO. Es letal en grandes concentraciones (40%). El CO presenta una afinidad 200 veces mayor que el oxígeno por la Hb desplazándolo a este fácilmente produciendo hipoxia tisular, pero con una coloración cutánea normal (produce coloración sanguínea fuertemente roja) (Hb+CO). 10. Hemoglobina glucosilada: presente en patologías como la diabetes , resulta de ka unión de la Hb con carbohidratos libres unidos a cadenas carbonadas con funciones ácidas en el carbono 3 y 4. 11. Tambien hay hemoglobinas de los tipos : Gower 1, Gower 2 y Pórtland. Estas sólo están presentes en el embrión. Son valores de referencia : Para Hombres: 16+/- 2,0. Para Mujeres: 14+/- 2,0
- 11. ANEMIA DE CÉLULAS FALCIFORMES (DREPANOCITOSIS, HEMOGLOBINOSIS S) ¿QUE ES LA ANEMIA DREPANOCITICA? La anemia drepanocítica o de células falciformes es un trastorno hereditario proteína de la sangre caracterizado por una anomalía de la hemoglobina (presente en los glóbulos rojos cuya función es transportar oxígeno a los tejidos del cuerpo). La anemia drepanocítica compromete los glóbulos rojos, o hemoglobina, y su capacidad de transportar oxígeno. Las células normales de hemoglobina son lisas, redondas y flexibles, como la letra “O”, por lo que se pueden desplazar fácilmente por los vasos del cuerpo. Por el contrario,las células falciformes (drepanocitos) de hemoglobina son rígidas y pegajosas,y adoptan la forma de hoz o letra “C” cuando pierden el oxígeno. Estas células falciformes tienden a aglutinarse y no pueden moverse fácilmente a través de los vasos sanguíneos. Las aglutinaciones producen un bloqueo y detienen el movimiento de la sangre normal y sana que transporta oxígeno. Este bloqueo es el que provoca las dolorosas y nocivas complicaciones de la anemia drepanocítica. Las células falciformes sólo viven 10 – 20 días, mientras que la hemoglobina normal puede vivir hasta 120 días. Además,las células falciformes corren riesgo de ser destruidas porel bazo debido a su forma y rigidez. El bazo es un órgano que ayuda a filtrar la sangre de infecciones y las células falciformes se atascan en ese filtro y mueren. Debido a la reducción del número de células de hemoglobina que circulan en el cuerpo, una persona con anemia drepanocítica padece de anemia crónica. El bazo también sufre daños, ya que las células falciformes bloquean las células sanas que transportan oxígeno. Sin un bazo que funcione normalmente, estas personas tienen mayores probabilidades de contraer infecciones. Los bebes y niños pequeños corren riesgo de contraer infecciones que pueden resultar mortales.
- 12. Las variedades comunes del gen de la celula falciforme son: 1. Rasgo drepanocítico:El niño es portadordelgen defectuoso, HbS, pero también tiene algo de hemoglobina normal, HbA (HbAS).Los niños que tienen el rasgo drepanocítico normalmente no presentan ningún síntoma de la enfermedad. Puede producirse una anemia leve. En situaciones estresantes, intensas, de cansancio extremo, de hipoxia (bajo nivel de oxígeno) y o de infección severa puede producirse el bloqueo de la hemoglobina defectuosa y esto puede provocar algunas complicaciones asociadas con la anemia drepanocítica. 2. Anemia drepanocítica: La mayoría o toda la hemoglobina normal (HbA) del niño está cambiada por hemoglobina falciforme (HbS), esto se denomina HbSS. Es la forma más común y más severa de las variedades de células falciformes. Estos niños padecen una serie de complicaciones por causa de la forma y el espesor de dichas células. La anemia severa y crónica es también una característica común de los niños que tienen HbSS 3. Anemia Drepanocítica- de hemoglobina C: El niño tiene HbS y HbC. Esto se denomina HbSC. La hemoglobina C genera el desarrollo de glóbulos rojos, denominados dianocitos. Si la persona tiene un poco de hemoglobina C y de hemoglobina normal, no tendrá ningún síntoma de anemia. Sin embargo, si la hemoglobina falciforme S se combina con el dianocito, puede producirse alguna anemia de leve a moderada. Estos niños a menudo sufren de algunas complicaciones asociadas con la HbSS, o anemia drepanocítica, pero en un grado más leve. Las crisis vasooclusivas (bloqueo del flujo sanguíneo por que las células falciformes se atascan en los vasos sanguíneos), el daño a organos a causa de bloqueos repetidos y anemia,y un alto riesgo de infección son rasgos similares de la HbSS y la HbSC. 4. Anemia Drepanocítica- de hemoglobina E: Esta variedad es similar a la anemia drepanocítica tipo C excepto en que se ha sustituido un elemento en la molécula de hemoglobina. Esta variedad se ve a menudo con las poblaciones del sudeste asiático. Algunos niños que tienen la enfermedad de hemoglobina E no presentan síntomas. Sin embargo, en determinadas condiciones, como cansancio extremo, hipoxia, infección grave y, o ferropenia (defiencia de hierro) puede desarrollarse anemia leve a moderada. 5. Hemoglobina S – beta talasemia: consiste en la herencia de los dos genes el de las células falciformes y el de la talasemia. El trastorno produce síntomas de anemia moderada y muchas de las mismas patologías asociadas con la anemia drepanocítica. Si bien este trastorno suele presentarsíntomas más leves que la anemia drepanocítica, tambien puede producir exacerbaciones tan graves como las de esta última.
- 13. Todas las formas de anemia drepanocítica pueden presentar las complicaciones asociadas con la enfermedad. A QUIENES AFECTA LA ANEMIA DREPANOCÍTICA? La anemia drepanocítica afecta principalmente a las personas de ascendencia africana y a los hispanos del caribe, pero tambien se ha encontrado el rasgo en descendientes de nativos americanos o proveniente de Medio Oriente, India, América Latina y el Mediterráneo. Se calcula que más de 70,000 personas en Estados Unidos padecen esta enfermedad.En el mundo millones de personas sufren las complicaciones de la anemia drepanocítica. QUÉ CAUSA LA ANEMIA DREPANOCÍTICA? La anemia drepanocítica es una enfermedad hereditaria causada por una mutación genética.Los genes se encuentran en unas estructuras de las células de nuestro cuerpo denominadas “cromosomas”. Normalmente hay 46 cromosomas en total (23 pares) en cada célula del cuerpo. El par 11 de cromosomas contiene un gen responsable de la producción de hemoglobina normal. Una mutación o error en este gen es lo que causa la anemia drepanocítica. Se cree que esta mutación se ha originado en las regiones del mundo donde la malaria era común, ya que las personas que presentan el rasgo de anemia drepanocítica no contraen malaria. El rasgo drepanocítico de hecho los protege del parásito que causa la malaria (transmitida por los mosquitos). La malaria es más frecuente en áfrica y en la zona del Mediterráneo europeo. La anemia drepanocítica (HbSS) es una enfermedad genética. Un bebe nace con anemia drepanocítica únicamente se hereda dos genes HbS, uno de su madre y el otro de su padre.
- 14. Las personas que sólo tienen un gen HbS están sanas, y se dice que son “portadoras” de la enfermedad. También se puede decir que tienen el rasgo drepanocítico. Un portador tienen mayores probabilidades de tener un bebé con anemia drepanocítica. Este tipo de herencia se denomina autonómica recesiva. “Autosómica” significa que el gen está en uno de los primeros 22 pares de cromosomas que no determina elgénero, por lo que la enfermedad afecta por igual a hombre y mujeres. “Recesiva” significa que son necesarias dos copias del gen, una heredada de cada progenitor, para padecer la enfermedad. Niños con anemia drepanocítica = S S (uno cada cuatro o 25%) Niños portadores delgen,al igual que sus padres = A S S A (dos cada cuatro, o 50 % presentan el rasgo drepanocítico). Niños que no heredan el gen de ningún progenitor: A A (uno cada cuatro,o 25%) Una vez que los padres han tenido un hijo con anemia drepanocítica, las probabilidades de que tengan otro hijo con este trastorno son de una en cuatro (25%). Esto significa que hay un 75% de probabilidadesque otro hijo no presente anemia drepanocítica. También hay un 50% de probabilidades de que un niño nazaca con el rasgo drepanocítico, al igual que los padres. ¿CUÁLES SON LOS SÍNTOMAS DE LA ANEMIA DREPANOCÍTICA? Anemia. En la anemia drepanocítica se producen glóbulos rojos pero después se deforman y adoptan la forma de hoz, l oque provoca que los eritrocitos pierdan su capacidad de transportar oxígeno. El cuerpo posteriormente se deshidrata o desarrolla fiebre. La forma de hoz hace a las células rígidas y pegajosas , en consecuencia quedan atascadas en los vasos sanguíneos,son destruidas por le bazo o simplemente mueren a causa de su función anormal. La disminución de los glóbulos rojos causa anemia. La anemia grave puede provocar que un niño se vuelva pálidoy se canse y dificulta la capacidad de su organismo para transportar oxígeno a los tejidos.
- 15. La anemia crónica puede retardarla curación así como también elcrecimiento y el desarrollo normales. Crisis de dolor o crisis de bloqueo – cuando el flujo sanguíneo se bloquea en una zona por que las células falciformes se han quedado atascadas en el vaso sanguíneo. Tambien se las denomina crisis vasooclusivas. El dolor puede presentarse en cualquier zona del cuerpo, pero lo más frecuente es que se sienta en el tórax o las extremidades. En los bebes y niños menores de 3 años se puede presentar inflamación dolorosa de los dedos de las manos y de los pies (dactilitis). El priapismo es un bloqueo doloroso que se presenta en el pene. Cualquier interrupción en el flujo sanguíneo al cuerpo puede provocar dolor , inflamación y posible muerte del tejido adyacente que no recibe la suficiente cantidad de sangre ni de oxígeno. Síndrome agudo de tórax – el bloqueo se sitúa en el tórax. Este tipo de complicación de la anemia de células falciformes puede ser fatal. A menudo se produce repentinamente cuando elcuerpo está bajo el estrés de una infección, fiebre o deshidratación. Las células falciformes se aglutinan y bloquean el flujo de oxígeno en los diminutos vasos pulmonares. Se parece a la neumonía y puede incluir fiebre, dolor y tos violenta. Múltiples episodios del síndrome agudo de tórax pueden causar daño permanente en los pulmones. Secuestro esplénico (acumulación)- crisis resultado de la acumulación de células falciformes en el bazo.Eso puede producir una disminución repentina de hemoglobina y poneren peligro la vida si no se trata rápidamente. El bazo también puede aumentarde tamaño y dolor como consecuencia del aumento del volumen de sangre. Después de episodios repetidos de secuestro esplénico, se forman cicatrices en el bazo, el cual queda permanentemente dañado. Alrededor de los 8 años, el bazo de la mayoría de los niños ya no funciona, ya sea por que se lo ha extraído quirúrgicamente o debido a la repetición de episodios de secuestro esplénico. El riesgo de infección es una preocupación de importancia en los niños con deficiencia esplénica.la
- 16. infección es la principal causa de muerte en los niños menores de 5 años en esta población. Accidente Cerebrovascular - otra complicación repentina y severa de los niños que tienen anemia drepanocítica. Las células deformadas pueden bloquear los principales vasos sanguíneos que aportan oxígeno al cerebro. Cualquier interrupción del flujo sanguíneo al cerebro puede producir un deterioro neurológico devastador. El niño que ha tenido un accidente cerebrovascular , tiene un 60% más de probabilidades de tener un segundo y un tercer episodio. Ictericia o color amarillento de la piel, los ojos y la mucosa bucal- señal y síntoma comunes de la anemia drepanocítica. Las células falciformes no viven tanto tiempo como los glóbulos rojos normales y, por lo tanto mueren a una velocidad más rápida que la capacidad de filtración del hígado. La bilirrubina de estas células degradadas se acumula en el sistema causando ictericia. Todos y cada uno de los órganos principales se ven afectados por la anemia drepanocítica. El hígado, el corazón, los riñones, la vesícula biliar, los ojos, los huesos y las articulaciones pueden sufrir daño como consecuencia de la función anormal de las células falciformes y su incapacidad de fluir correctamente a través de los pequeños vasos sanguíneos. Los problemas pueden incluir: 1. Aumento de las infecciones 2. ülceras en las piernas 3. Daño óseo 4. Colelitiasis prematura (cálculos biliares) 5. Daño renal y pérdida de agua corporal en la orina 6. Daño ocular Los síntomas de la anemia drepanocítica pueden parecerse a los de otros trastornos de la sangre o problemas médicos. ¿CÓMO SE DIAGNOSTICA LA ANEMIA DREPANOCITICA? TRATAMIENTO DE LA ANEMIA DREPANOCITICA El tratamiento específico se basa en lo siguiente: La edad de su hijo, su estado general de salud y su antecedentes médicos. La gravedad de la enfermedad.
- 17. La tolerancia de su hijo a determinados medicamentos, procedimientos o terapias. Sus expectativas para la evolución de la enfermedad El Tx incluye: 1. Analgésicos para las crisis de la anemiadrepanocítica 2. Beber mucho agua diariamente de 9 – 10 vasos o por vía IV para prevenir las crisis 3. Transfusiones de sangre para evitar la anemia y el accidente cerebrovascular se utilizan también par diluir la HbS con hemoglobina normal con el fin de tratar el dolor crónico, el síndrome agudo de tórax, el secuestro esplénico y otras emergencias. 4. Penicilina para prevenir infecciones. 5. Ácido fólico para ayudar a prevenir la anemia grave 6. Hidroxicarbamida un medicamento recientemente desarrollado que puede ayudar a reducir la frecuencia de las crisis de dolor y del síndrome agudo de tórax; también puede ayudar a disminuir la necesidad de frecuentes transfusiones sanguíneas.Se desconocen los efectos a largo plazo del medicamento. 7. Transplantede médula ósea ha sido eficaz para curar a algunos niños con anemia drepanocítica; la decisión de someterse a este procedimiento se funda en la gravedad de la ptología y en la disponibilidad de un donante adecuado. CONSIDERACIONES GENERALES: Enfermedad de transmisión recesiva Esquemáticamente hay dos formas: Homocigoto S/S portador de un 70 a 80% de Hb S (forma más grave) y heterozigotos A/S portadoresde un 20 a 40% de Hb S (con manifestaciones atenuadas de la enfermedad) Existen otras formas asocidad a varios tipos de anomalías genéticas: 1. Asociación de hemoglobinosis S con Hemoglobinosis C y de hemoglobinosis S y déficit de G6PD. La forma de los hematíes cambia con el descenso de la presión parcial de O2. De biconcavos las células se hacen más o menos espiculadasy con forma de hoz (falciformación). Es en principio reversible después irreversible, acompañandose de alteraciones rheológicas de los hematíes con microtrombósis e infartos
- 18. titulares; y también hay alteraciones de la membrana que producen una hemolisis. En las formas greaves se producen crisis vaso-oclusivas (dolor osteoarticular en manos y pies, lumbar, torácico, abdominal), por microtrombósis. Tambien pueden aparecer accidentes cerebrovasculares. Infecciones frecuentes, con trastornos respiratorios. Fibrosis Intersticial pulmonar. Han sido descritas lesiones glomerulares. Cardiomiopatía. CONSIDERACIONES ANESTÉSICAS: Estudio preoperatorio 1. Búsqueda sistemática de células falciformes. 2. Búsqueda de un déficit de G6PD en la raza negra y con antecedentes de crisis hemolíticas. 3. Examen cardíaco completo y de la función pulmonar . 4. Búsqueda de focos infecciosos o parasitarios. 5. Para algunos la antibioterapia es obligatoria. MANEJO DE ENFERMEDADES CON RASGOS DE CÉLULAS FALCIFORMES (Hb AS): 1. Mantener la oxigenación 2. Prevenir la hipotermia 3. Prevenir deshidratación 4. Precaución con el uso prolongado del torniquete. 5. En pacientes con niveles altos de HbS y en intervenciones mayores debería considerarse una exanguinotransfusión preoperatorio. MANEJO DE ENFERMEDAD DE CÉLULAS FALCIFORMES (Hb SS, HbSC, Hb S,beta-talasemia) 1. Anemia (Hb 5 – 8 g/dl) 2. Cor pulmonales alteraciones renales, sobrecarga de hierro, enfermedad cerebrovascular, susceptibilidad a la infección bacteriana. 3. Normalmente es necesario el exanguinotransfusión. Mantener niveles de Hb A >/= 50% y la Hb en 10 gm/dL. 4. Alcalinización preoperatorio. 5. Mantenimiento de la temperatura. 6. Mantenimiento de la hidratación 7. Mantener oxigenación. 8. Atención al posicionamiento en la mesa de operaciones por el extasis venoso. 9. Los torniquetes usarlos solo si es absolutamente necesario
- 19. 10.En cirugía mayor monitorización de la saturación venosa mixta. 11.Atención al postoperatorio puede desencadenarse una crisis Una mujer de raza negra de 24 años de edad con antecedente familiar de anemia de células falciformes hereditaria, se presenta con dolor abdominal y es programada para practicar Colecistectomía. La paciente cree que padece anemia de células falciformes. ¿Qué es la anemia de células falciformes? Es una anemia hemolítica hereditaria causada por la formación de una hemoglobina normal del adulto (Hb-A) sólo en la sustitución de valina en lugar de ácido glutámico en la posición 6 de la cadena beta. Funcionalmente, la hemoglobina falciforme tiene menor afinidad por el oxígeno (P50 = 31 mmHg), así como solubilidad disminuida. Después de desoxigenarse, la Hb S polimeriza con facilidad y se precipita en el interior de los eritrocitos, lo que hace que adquieran aspecto de hoz. Los pacientes producen cantidades variables (2 – 20%) de hemoglobina fetal (Hb F). Es probable que las células con cantidades grandes de Hb F estén un tanto protegidas de la formación falciforme. La formación y destrucción continua de células falciformes causa anemia. Los hematocrito típicos son de 18 a 30% causa de hemólisis intravascular. La supervivencia de los eritrocitos se reduce a 10 a 15 días, comparada con hasta 120 días en individuos normales. ¿CUÁL ES LA DIFERENCIA ENTRE LA ANEMIA DE CÉLULAS FALCIFORMES Y EL RASGO DE CÉLULAS FALCIFORMES? Cuando el defecto genético de la hemoglobina deladulto se deriva tanto de cromosomas de la madre como del padre (Num. 11), el paciente es homocigoto para Hb S y tiene anemia de células falciformes (Hb SS). Cuando sólo un cromosoma tiene el gen falciforme, el paciente es heterocigotoy tiene rasgo falciforme producen cantidades variables de Hb
- 20. A (55- 60%) y Hb S (35 – 40%).A diferencia de los que tienen Hb SS, no suelen ser anémicos, son asintomáticos y tienen un período normal de vida. La formación falciforme se produce sólo en hipoxemia intensa o en estados de flujo bajo. La formación falciforme tiene un potencial especial para producirse en la médula renal; de hecho,muchos pacientes con rasgo de células falciformes tienen deterioro en la capacidad de concentración renal. Se informa que algunos pacientes con Hb AS tienen infartos renales medulares, esplénicos y pulmonares. ¿CUÁL ES LA FRECUENCIA DEL GEN DE CÉLULAS FALCIFORMES EN LOS ESTADOUNIDENSES DE RAZA NEGRA? La anemia de células falciformes es sobre todo una enfermedad de personas de raza negra con antecesores de África Central. Cerca de 0.2 – 0.5% de los individuos que pertenecen a este grupo es homocigoto para el gen falciforme, mientras que alrededor de 8 – 10% es heterocigoto. La anemia de células falciformes se encuentra menos en pacientes con antecesores mediterráneos. ¿CUÁL ES LA FISIOPATOLOGÍA DE LA ANEMIA DE CÉLULAS FALCIFORMES? Los padecimientos que favorecen la formación de desoxihemoglobina – como hipoxemia, acidósis, hipertonicidad o deshidratación intracelular, aumento en las concentraciones de 2,3 DPG, o incremento en la temperatura – pueden precipitar formación falciforme en pacientes con Hb SS. La hipotermia tambien es perjudicial debido a la vasoconstricción coexistente. La polimerización intracelular de Hb S produce distorsión de los eritrocitos, que son menos plegables y “más pegajosos”, y aumenta la viscosidad de la sangre. La formación falciforme puede ser reversible al inicio, pero al final se vuelve irreversible en algunas células. La formación de agregados de eritrocitos en los capilares obstruye la microcirculación en los tejidos.
- 21. Se establece un círculo vicioso en el cual la estasis circulatoria causa hipoxia localizada, que a su vez origina más formación falciforme. ¿ QUE SÍNTOMAS PRESENTA EL PACIENTE CON ANEMIA DE CÉLULAS FALCIFORMES? Los pacientes con Hb SS suelen desarrollar suelen desarrollar síntomas durante la infancia, cuando las concentraciones de hemoglobina fetal (Hb F) declinan de modo apreciable. La enfermedad se caracteriza tanto por crisis episódicas agudas como por manifetaciones crónicas y progresivas. MANIFESTACIONES DELA ANEMIA DE CÉLULAS FALCIFORMES NEUROLÓGICAS Evento vascular cerebral Hemorragia Subaracnoidea Coma Convulsiones OCULARES Hemorragia del humor vítreo Infarto retiniano Retinopatía proliferativa Desprendimiento de retina PULMONARES Aumento de cortocircuitos intrapulmonares Pleuritis Infecciones pulmonares recurrentes Infartos pulmonares
- 22. CARDIOVASCULARES Insuficiencia Cardíaca Congestiva Cor pulmonale Pericarditis Infarto Miocardico DIGESTIVAS Colelitiasis (Cálculos pigmentados) Colecistitis Infartos hepáticos Abcesos hepáticos Fibrosis Hepática HEMATOLÓGICAS Anemia Anemia Aplásica Infecciones recurrentes Infarto Esplénico Secuestro Esplénico Asplenia funcional GENITOURINARIAS Hematuria Necrosis papilar renal Deterioro de la capacidad de concentración renal (isostenuria) Síndrome nefrótico Insuficiencia renal
- 23. Priapismo ESQUELÉTICAS Sinovitis Artritis Necrósis aséptica de la cabeza femoral Infartos óseos pequeños en manos y pies (dactilitus) Vertebras biconcavas Osteomielitis CUTÁNEAS Úlceras crónicas Los niños sufren retardo del crecimiento e infecciones recurrentes. Los infartos esplénicos recurrentes conducen a atrofia esplénica y asplenismo funcional hacia la adolescencia. Los pacientes suelen morir a causa de infecciones recurrentes o insuficiencia renal. A menudo,las crisis se precipitan por infecciones, clima frío, deshidratación u otras formas de estrés. Las crisis se dividen en 3 tipos: 1. Crisis Vaooclusivas: Dependiendo de los vasos afectados, estos episodios agudos producen microinfartos o macroinfartos. Se considera que la mayor parte de las crisis dolorosas se debe a microinfartos en los diversos tejidos. Clínicamente, hay dolor abdominal agudo,torácico, de espalda o articular. Es difícil diferenciarentre causas quirúrgicas y no quirúrgicas del dolor abdominal. La mayoría de los pacientes forma cálculos biliares pigmentados hacia la edad adulta, y muchos presentan colecistitis aguda.
- 24. Los fenómenos vasooclusivos en vasos más grandes pueden producir trombosis que causan infartos esplénicos, cerebrales, pulmonares, hepáticos, renales y con menos frecuencia miocárdicos. 2. Crisis Aplásica 3. Crisis de Secuestro Esplénico TRANSFUSIÓN EN LA ANEMIA DE CELULAS FALCIFORMES O ANEMIA DREPANOCITICA (siempre con filtro de leucocitos) En general toleran bien HTO bajos, por lo que no hay una indicación clara de transfusión con Hb 5 gm/dL. Cantidad de concentrado de hematíes a transfundir (intentar no superar una Hb 10 gm/dL ó HTO 30%). INDICACIONES DE TRANSFUSIÓN SIMPLE AISLADA Hb 5 gm/Dl ó HTO 15%
- 25. MORFOLOGIA POIQUILOCITOSIS:–CELULA DESIGUAL,NOCIRCULAR, PERIFORME, ABOTAGADA. –A. HIPOCROMICA, S. MILEOPROLIFERATIVO, CA OSEO. •PLANOCITOS O LEPTOCITOS:–CELULASDELGADAS, APLANADAS. –A. HIPOCROMICAS. •ANULOCITOS:–CELULA FORMA DE ANILLO, EXAGERADA DEPRECION CENTRAL. –A. HIPOCROMICA. ESFEROCITOS –CELULA EN FORMA DE BOLA, REDONDA,PEQUEÑAS, HIPERCROMICAS. –A. HEMOLITICAS. •OVALOCITOS –ELIPTOCITO–ALARGADOS, EN FORMA DE HUEVO. –A. FERROPENICA, MEGALOCITOS.
- 26. •DACRIOCITOS -TEARDROP–ENFORMA DE LAGRIMA ORAQUETA –A. FERROPENICA OMEGALOBLASTICA, MIELOFIBROSIS, TRAS. HEMATOPOYETICO ESQUISTOCITOSIS -O FRACMENTOCITOSIS–FRACMENTOSDE CELULASEN FORMA DE TRIANGULO, YELMO, CASCO, LAGRIMA. –HEMOLISISMECANICA, A. MICROANGIOPATICA OPOR PROTESIS VALVULAR, PURPURA TROMBOTICA TROMBOCITOPENICA. •DIANOCITOS O CELULAS DIANA <<TARGET CELLS>> –ZONA CENTRALABULTADA HIPERCROMICOS, HALOPALIDO, ANILLO PERIFERICONORMAL –A. HEMOLITICA •DREPANOCITOS OCELULAS FALCIFORMES <<SICKLE CELLS>> –EN FORMA DE GUADAÑA OSEMILUNA. –HEMOGLINOPATIASS ACANTOCITOSIS –BURR CELLS–CELULASERIZADASCON ESPINASO ESPICULAS.–ABETALIPOPROTEINEMIAS(CON RETINITIS PIGMENTARIA Y ATAXIA)OLA NEUROACANTOSIS(CON COREA), TAMBIEN SE A DESCRITOEN LAS A. HEMOLITICASMICRO ANGIOPATICAS, PTT, DE PIRUVATOQUINASA–NOCONFUNDIRLA EQUINOCITOS DE PROMINENCIASCORTAS –EN CIRROSISY S. ZIEVE.•ESTOMATOCITOS–DEPRECION CENTRALEN FORMA DE BOCA O RANURA–POR CARENCIA DE ATPASA ,A. HEMOLITICA HEREDITARIA, ALCOHOLISMO, SATURNISMOY TALASEMIA. CROMEMIA 1. NORMO CRONEMIA:Coloracion normal de hematies. 2. HIPOCRONEMIA:Hematies palidos propios de la anemia ferropenica. 3. HIPERCRONEMIA:Coloracion superior a lo normal, ej.: La esferocitosis. 4. ANISOCROMIA:Coexistencia de eritrocitos normocromicos e hipercromicos en a. sideropenicas en tto o despues de transfucion 5. POLICROMATOFILA:Hematies grisazulacios entre los normales. reticulocitos propios de anemias regenerativas TAMAÑO 1. NORMOCITOSIS: Hematies de tamaño igual 2. MACROCITOSIS: Tamaño mayor de lo normal. en alcoholismo, anemia megaloblastica (perniciosa) 3. MICROCITOSIS: Tamaño inferior a lo normal. anemias ferropenicas, esferocitosis, talacemia minor sinanemia. 4. ANISOCITOSIS: Desigual entre si, en la hepatopatia cirrotica sangrante y enf. del intestino delgado
- 27. BILIRRUBINA SERICA:Bilirrubina Total (Conjugada y No Conjugada) es inf a 1.5 mg/dL refleja el equilibrio entre la producción y la excreción biliar. ICTERICIA PRESENTE Bilirrubina Total de 3 mg/dl Hiperbilirrubinemia Conjugada > 50% + Urobilinógeno urinario refleja DISFUNCIÓN HEPATOCELULAR, COLESTASIS INTRAHEPÁTICA U OBSTRUCCIÓN BILIAR EXTRAHEPÁTICA Hiperbilirrubinemia No Conjugada Hemólisis o defectos congénitos adquiridos en la conjugación de la bilirrubina. La bilirrubina No conjugada no es tóxica para las célular AMINOTRANFERASAS SÉRICAS: Enzimas que se liberan por lesión o muerte hepatocelular ASPARTATO AMINOTRANFERASA SERICA (AST ó TGO): Pte en el hígado, corazón,músculo esquelético y riñones ALANINA AMINOTRANFERASA SÉRICA (ALT ó TGP): Se localiza en el hígado y es más específica de disfunción hepática Concentraciones normales de ALT y AST son de 35 – 45 unidades/L, elevaciones de 300 unidades se observan en COLESTASIS O ENF METASTASICA DEL HÍGADO SON UTILES EN ENF HEPATICAS AGUDAS POR SOBREDOSIS DE MEDICAMENTOS, LESIÓN ISQUÉMICA Y HEPATITIS FULMINANTE FOSTASA ALCALINA: Se produce en el hígado, hueso, intestino delgado, riñones y placenta, la mayor parte se deriva del hueso pero en caso de obstrucción biliar se sintetiza más FA hepática y se libera a la circulación y se excreta por la bilis Actividad normal: 25 – 85 U/L Elevaciones del doble de lo normal se ven en las lesiones hepatocelular o enf metastasica del hígado. Concentraciones más elevadas indican colestasis intrahepática u obstrucción biliar ALBUMINA SÉRICA: Concentración normal: 3.5 – 5.5 g/dL Al principìo de una enf hepática aguda puede ser normal Valores de 2.5 gm/dL indican enf hepática crónica, stréss agudo o desnutrición grave. Incremento en las pérdidas de albúmina por orina (Síndrome Nefrótico) o vías digestivas ( Enteropatía con desgastes de proteínas) tambien produce hipoalbuminemia AMONÍACO SANGUÍNEO:
- 28. Elevación importante de las concentraciones de amoníaco en sangre refleja alteración en la síntesis hepática de urea. Concentraciones normales de amoníaco en sangre son de 47 – 65 mmol/L (80 – 110 mg/dL) Su elevación indica daño hepatocelular grave, Aunque no existe relación de amoníaco arterial y la encefalopatía hepática TIEMPO DE PROTROMBINA: TP es de 11 – 14 segundos Mide la actividad del fibrinógeno, protrombina y los factores V, VII y X. La vida media corta del factor VII ( 4 – 6 horas) hace que el tiempo de protrombina sea útil en lla evaluación sintética del hígado en px con enf hepática aguda a crónica. CLASIFICACIÓN DE LAS ANEMIAS A.Según volumen corpuscular medio (82-92fl) – Anemia microcítica (VCM < 80 ft) – Anemia normocítica ( VCM 80-96 ft) – Anemia macrocítica ( VCM > 96 ft) B.Según recuento de reticulocitos – Anemia regenerativa > 150 mil reticulocitos – Anemia arregenerativa < 150 mil reticulocitos ANEMIA MEGALOBLÁSTICA Las anemias megaloblásticas tienen en común ciertas alteraciones en la forma y maduración de los hematíes que son de valor diagnóstico. En cualquier estadío del desarrollo, los hematíes son mayores de lo normal y muestran disposición laxa y dispersa de la cromatina nuclear, así como una asincronía entre la maduración del núcleo y el
- 29. citoplasma; el retraso de la maduración nuclear es más evidente a medida que la célula presenta un mayor número de divisiones. La forma megaloblástica se deben a un déficit de ácido fólico, vitamina B12 o de ambos. Las dos sustancias son cofactores indispensables en la síntesis de las núcleoproteínas y su déficit conlleva a una síntesis anómala del ADN y, en menor medida, del ARN y las proteínas. DATOS DE LABORATORIO: La eritropoyesis ineficaz se debe a la detención del desarrollo o a la muerte prematura de las células de la médula ósea. En la sangre periférica, los hematíes son mayores (aumento del volumen corpuscular medio, VCM) y con frecuencia de forma ovalada, aparecen neutrófilos hipersegmentados y también se pueden observar plaquetas gigantes. En la médula ósea los hematíes megaloblásticos nucleados tardíos pueden presentar una cantidad suficiente de hemoglobina, pero todavía retienen su núcleo inmaduro en vez del habitual grumo de cromatina. En la médula ósea también se pueden observar metamielocitos gigantes y formas en banda La anemia es macrocítica (VCM > 100fL). El tamaño y la forma de los hematíes varía con frecuencia. Los reticulocitos escasean, pero es frecuente observar hematíes nucleados de forma megaloblástica en la sangre. Leucopènia, especialmente en los déficit de larga evolución Trombocitopenia , especialmente en los déficit de larga evolución Los neutrófilos son hipersegmentados; más del 5% de los neutrofilos tienen 5 o más lobulaciones del núcleo. Los niveles séricos normales de ácido fólico son de 5 – 20 ng/mL; cuando hay déficit se observan valores por debajo de 3 ng/mL. Los niveles de folato
- 30. HEPATOPATIAS Y ANESTESIA Se refiere a cualquiera de las enfermedades del hígado Se dividen en 2 grandes grupos: 1. Afección del parénquima: Hepatitis Viral Aguda y Crónica, Cirrosis Hepática con o sin hipertensión portal 2. Enfermedad Hepatobiliar: Colestasis y obstrucción de las vías biliares Enfermedad Hepática Crónica 1. Metabólicas Enf de Wilson Deficiencia de antitripsina alfa 1 Hemocromatosis Amiloidosis 2. Drogas que inducen reacciones idiosincráticas 3. Hepatitis granulomatosa 4. Sarcoidosis 5. Hepatitis Crónica Persistente 6. Hepatitis Crónica Activa 7. Hepatitis Alcohólica Cirrosis de Laenec 8. Cirrosis 9. Tumores 10. Carcinoma Hepatocelular 11. Enf Metastásica 12. Desordenes Linfoproliferativos CAUSAS COMUNES DE ICTERICIA CLASIFICACIÓN NO HEPÁTICAS 1. Hemólisis 2. Sd de Gilbert
- 31. INTRAHEPATICAS Ó DE PARÉNQUIMA 1. Enf Infecciosas Hepatitis A,B C y No A , No B 2. Relacionadas al SIDA CMV Herpes TB Parasitósis Sd de Reyé 3. Trauma 4. Sd de Budd Chiari OBSTRUCTIVA 1. Obstrucción Metabólica Sd de Rotor Sd Dubin- Jhonson 2. Lesión Congénita del tracto biliar Aplasia Biliar Enf Poliquística 3. Obstrucción Extrahepática Colecistitis Coledocolitiásis Carcinoma del Páncreas Conlangiocarcinoma Colangitis esclerosante HEPATITIS VIRAL Se debe a infección viral( Virus de Hepatitis A,B ó C), reacción a fármacos o exposición de hepatotoxinas. Repta lesión hepatocelular aguda con grados de necrosis celular Manifestaciones Clx dependen de la intensidad de reaccion inflamatoria y cantidad de necrósis HEPATITIS INDUCIDA POR FÁRMACOS
- 32. Se debe a toxicidad directa, dep de la dosis de un medicamento (o un metabolito), por una reacción medicamentosa idiosincrática o por combinación de ambas. Evolución similar a la hepatitis viral dificil dx Hepatitis alcohólica es la más frect. HEPATITIS AGUDA HEPATITIS INDUCIDA POR FÁRMACOS FARMACOS Y SUSTANCIAS QUE SE RELACIONAN CON HEPATITIS TOXICA IDIOSINCRACIA TOXICA E IDIOSINCRACICA COLESTACICA PRIMARIA Alcohol Acetaminofen Salicilatos Tetraciclinas Halotano Fenitoina Sulfonamidas Rifampicina Indometacina Metildopa Isoniacida Amiodarona Anticonceptivos orales Metamizol Esteroides anabólicos Estolato de Eritromicina CONSIDERACIONES PREOPERATORIAS Cirugía electiva se pospone hepatitis aguda resuelta reflejadas en pruebas de funcionamiento hepático Cirugía de urgencia, evaluación anestésica de causa y grado de deterioro hepático mortalidad 10% en laparotomía y morbilidad 12% perioperatorias Información: exposición a fármacos, ingestión de alcohol, abuso de drogas, transfusiones recientes y anestésicos previos. Hepatitis > riesgo de Insuficiencia Hepática como encefalopatía, cuagulopatía o sd hepatorrenal. Ex. De Lab.:BUN, electrolitos, Creatinina, glucosa, TGO, TGP, Bilirrubina, FA y albúmina sérica,TP y recuento de plaquetas, HBsAg. Cuantificación de alcohol en sangre Estado mental signos compatibles de intoxicación etílica alteración hepática grave. desorientación en los alcohólicos son signos de intoxicación aguda, el temblor e irritabilidad indican supresión. Hipopotasemia y alcalosis metabólicavómitos. Hipomagnesemia en alcohólicos crónicos Arritmias. TGO y TGP no se relaciona con necrósis La ALT serica es mas elevada que la AST con excepción de la hepatitis alcohólica donde se produce lo contrario.
- 33. Bilirrubina y FA TPprolongación > 3 seg después de la admón de vitamina K disfunción hepática grave Hipoglucemia No hay hipoalbuminemia excepto en casos crónicos con DN grave o con Enf. Hepática Crónica Antec. De násuseas y vómitosCorregir deshidratación y alterac. Electrolíticas La vitamina K ó el PFC puede corregir cuagulopatía. No premedicar la exposición a medicamentos y evitar confundir con encefalopatía hepática BZD y tiamina indicadas en px alcohólicos con manifestaciones agudas de supresión. CONSIDERACIONES TRANSOPERATORIAS Objetivo del tx operatorio: Preservar la función hepática existente y evitar factores perjudiciales para el hígado. SELECCIÓN DE FÁRMACOS Y DOSIFICACIÓN INDIVIDUALIZADA. Px con hepatitis viral ptan en la sensibilidad del SNC a los anestésicos TODOS LOS ANESTÉSICOS SON DEPRESORES DEL SNC POR TANTO DEBE USARSE EL MENOR NÚMERO DE ESTOS. ANESTÉSICOS POR INHALACIÓN SON PREFERIBLES A LOS INTRAVENOSOS, YA QUE LA MAYOR PARTE DE ESTOS ÚLTIMOS DEPENDEN DEL HÍGADO PARA SU METABOLISMO O ELIMINACIÓN. Dosis estándar de agentes de inducción intravenosos, su acción es determinada por redistribución más que por el metabolismo o excreción. DURACIÓN PROLONGADA DE AGENTES IV CON DOSIS GRANDES O REPETIDAS , EN ESPECIAL OPIODES. ISOFLURANE es el agente volátil preferido por menor efecto sobre el flujo sanguíneo hepático. Evitar factores perjudiciales que reduzcan el flujo sanguíneo hepático: Hipotensión Activación hepática excesiva Presiones medias elevadas de las vías respiratorias durante la ventilación controlada. En ausencia de coagulopatía Anestesia regional pero evitando la hipotensión HEPATITIS CRÓNICA Se clasifican en 1 – 3 síndromes diferentes con base en biopsia de hígado:
- 34. 1. Hepatitis Persistente Crónica 2. Hepatitis Lobulillar Crónica 3. Hepatitis activa Crónica HEPATITIS PERSISTENTE CRÓNICA: Inflamación hepática persistente de duración > 6 meses. Elevación de aminotransferasas séricas. Inflamación crónica de las vías portales con preservación de la arquitectura celular normal en la biopsia Progresa muy pocas veces a cirrosis Estado clínico, px se ptan como hepatitis aguda (por lo regular hepatitis B ó C) de curso prolongado, que al final resuelve HEPATITIS LOBULILLAR CRÓNICA: Hepatitis aguda que se resuelve, continuada con exacerbaciones recurrentes Focos de inflamación y necrósis en los lobulillos hepáticos, Pocas veces progresa a la cirrosis. HEPATITIS ACTIVA CRÓNICA: Inflamación hepática crónica con destrucción de la arquitectura celular normal (necrósis por partes) en la biopsia Cirrosis inicial de 20 – 50% ó se desarrolla al final. Consecuencia de hepatitis B ó C Otras causas (metildopa, oxifenisatina, isoniacida y nitrofurantoína) y trastornos inmunitarios como una predisposición genética. Síntomas: fatiga e ictericia recurrente, manifestac. Extrahepáticas como artritis yserositis. Px con enf progresiva manifestac de cirrosis. Pbas de Lab. : TGO y TGP Px sin infección crónica por hepatitis B o C responden favorablemente a inmunosupresores , se tratan con corticosteroides prolongadamente con azatioprina o sin ella. Estos px ptan manifestac autoinmunitarias: diabetes o tiroiditis, y tx corticosteroide prolongado TX anestésico igual que la hepatitis aguda Las causas de ascitis, como proceso de acumulacion de liq en cavidad peritoneal se pueden clasificar en:
- 35. Hepaticas; Hepatitis alcoholicas, cirrosis de cualquier etiologia, tumores primarios y secundarios, etc. Hematologicas: Sd de Budd Chiari Cardiovasculares: Pericarditis Constrictiva, ICC Peritoneales; Tuberculosis Neoplasicas; Carcinomatosis peritoneal, linfoma y SIDA CIRROSIS DEFINICION: Enf. Grave y progresiva que termina en Insuficiencia Hepática. ETIOLOGIA: 1. Alcohol (Cirrosis de Laennec)Causa más importante 2. Hepatitis Activa Crónica Cirrosis Posnecrótica 3. Inflamación u obstrucción biliar crónica Cirrosis biliar 4. Insufic. Cardíaca Congestiva Crónica Derecha Cirrosis Cardíaca 5. Hemocromatosis 6. Enf. De Wilson 7. Eficiencia de alfa1 antitripsina “SIN IMPORTAR LA CAUSA, LA NECRÓSIS DEL HEPATOCITO ES SEGUIDA POR FIBROSIS Y REGENERACIÓN NODULAR”. CLASIFICACIÓN DE CIRROSIS BASADA EN ASPECTOS ETIOLÓGICOS 1. Cirrosis por enf genéticas a. Galactosemia b. Enf de depósito de glucógeno c. Tirosinosis d. Intolerancia a la fructosa e. Deficienicia de alfa 1 antitripsina f. Talasemia y otras anemias g. Enf de Wilson h. Hemocromatosis i. Cirrosis biliar incompleta j. Telanguiectasias
- 36. k. abetalipoproteinemia l. Cirrósis química m. Secundaria a dano tóxico n. Cirrosis alcohólica o. Cirrosis Infecciosa p. Cirrosis por heaptitis viral q. Sifilis congénita r. Infecciones parasitarias s. Cirrosis nutricional t. Después de cirugía derivativa intestinal para tx de la obesidad u. Cirrosis biliar primaria v. Cirrosis Congestiva w. Cirrosis Criptogénica x. Cirrosis biliar secundaria HIPERTENSIÓN PORTAL Las causas de hipertension portal pueden clasificarse de acuerdo a su localizacion en: 1. Prehepaticas (Trombosis de la vena esplenica, por ejemplo) 2. Intrahepaticas ó de Parénquima (Hepatitis Viral Aguda y Crónica, Cirrosis Hepática con o sin hipertensión Portal Cirrosis,hepatocarcinoma) 3. Poshepaticas ( No necesariamente relacionadas con enf. Hepatica con Sd de Budd Chiari, Pericarditis Constrictiva) La distorsión de la arquitectura celular y vascular normal del hígado obstruye el flujo venoso portal y genera hipertensión portal, mientras el deterioro de las funciones sintéticas normales y otras funciones metabólicas diversas del hígado producen enf multisistémica. Los signos y sx no se relacionan con la intensidad de la enfermedad. Al principio las manifestaciones están ausentes, pero al final se desarrollan ictericia y ascitis en la mayoría de los px. Otros signos incluyen: angiomas arácneos, eritema palmar ,ginecomastia y esplenomegalia La cirrosis suele coexistir con 3 complicaciones principales: Varices hemorrágicas por la hipertensión portal
- 37. Retensión de líquidos intratable, bajo la forma de ascitis y síndrome hepatorrenal Encefalopatía o Coma Hepáticos Cerca del 10% de los px desarrolla un episodio de peritonitis bacteriana espontánea y algunos presentan al final carcinoma hepatocelular. Algunos producen fibrosis hepática sin necrósis hepatocelular ni regeneración nodular Ocasionan hipertensión portal y sus complicaciones (esquistosomiasis, fibrosis portal idiopática (Sd de Banti) y fibrósis hepática congénita) La obstrucción de las venas hepáticas o cava inferior (Sd de Budd Chiari ) esta se debe a trombósis venosa (estado de hipercuagulabilidad), un trombo tumoral (Carcinoma renal) ó enf. Oclusiva de las venas hepáticas sublobulares. Hipertensión Portal (> 10 mmHg) genera colaterales venosas portosistémicas extensas Se reconocen 4 sitios colaterales principales: 1. Gastroesofágico 2. Periumbilical 3. Retroperitoneal La HTP se evidencia antes de la intervención bajo la forma de venas dilatadas en la pared abdominal (cabeza de medusa) Hemorragia masiva por varices gastroesofágicas causa importante de morbilidad y mortalidad La pérdida aguda de sangre, el aumento en la carga de nitrógeno (por descomposición de la sangre en el tubo digestivo) precipita encefalopatía hepática. Endoscopia Procedimiento dx y terapéutico muy valioso. Es vital identificar el sitio de hemorragia ya que requieren tx distinto Tx de hemorragia varicosa es de soporte médico Pérdida Sanguínea se reemplaza con líq IV y hemoderivados TRATAMIENTO NO QUIRÚRGICO: Vasopresina 0.1 – 0.9 unidades/min IV, Somatostatina 250 mcg seguida por 250 cmg/h, Propranolol, taponamiento con Sonda de Segstaken- Blakemore o esclerosis endoscópica de las varices.
- 38. La vasopresina, somatostatina y Propranolol reducen la cantidad de sangre pérdida Dosis altas de vasopresina ocasionan ICC o Isquemia miocardica, pero en conjunto de Nitroglicerina IV disminuye la probabilidad Esclerosis endoscópica o la ligadura de las varicesEfectiva en el 90% de los sangrados. Derivaciones portosistémicas intrahepáticas transyugulares percutáneas reducen la hipertensión portal y la hemorragia pero aumenta la incidencia de encefalopatía. Cuando la hemorragia no se suspende se indica la cirugía de urgencia El riesgo quirúrgico se relaciona con el grado de deterioro hepático, según los signos clínicos y de laboratorio CLASIFICACIÓN DE CHILD Derivaciónes Px de riesgo bajo Ablación,sección esofágica y desvascularización gástrica Px de alto riesgo RIESGO QUIRURGICO QUE SE RELACIONA CON EL GRADO DE DETERIORO HEPÁTICO SEGÚN LOS SIGNOS CLINICOS Y DE LABORATORIO FACTOR DE RIESGO A B C Bilirrubina (mg/dl) <2 2 - 3 >3 Albúmina Sérica (g/dL) 3 – 5 3 - 3.5 3 Ascitis Ninguna Controlada Mal controlada Encefalopatía Ausente Mínima Coma TP Prolongado 2 2 – 3 3 Nutrición Excelente Buena Mala Ïndice de Mortalidad 2 - 5 10 50 VALORACION DE CHILD RIESGO CLINICO FACTORES MINIMA MODERADA SEVERA Encefalopatía Ninguna Provocada Grado 2 - 4 Ascitis Ninguna Controlada Incontrolable Nutrición Buena Buena Pobre Bilirrubina (mg/dl) 2 2-3 3 Bilirrubina (µmol/L) 34 34 - 50 50 TP Prolongado 2 2-3 3 Albúmina 3 - 5 3 – 3.5 3 Albúmina (g/L) 35 30 - 35 30
- 39. Anemia, trombocitopenia, leucopenia Causas de Anemia: pérdida de sangre, aumento de la destrucción de eritrocitos, supresión de la médula ósea y deficiencias de la nutrición. Esplenomegalia congestiva por HTTP Causa de Trombocitopenia y leucopenia Disminución en la síntesis hepática Deficiencia de factores de coagulación Aumento de la fibrinolisis por disminución de la depuración de los activadores del sistema fibrinolítico Contribuye a la coagulopatía Transfusiones preoperatorias en la carga de nitrógeno. Descomposición de proteínas por transfusiones excesivas Encefalopatía Corregir Coagulopatía antes de la operación. restituir los factores de coagulación como FPP y crioprecipitados , cifras menores de 100, 000/ μ ml de plaquetas y considerar la transfusión antes de la cirugía. Estado circulatorio hiperdinámico Aumento del GC Vasodilatación Periférica generalizada Cortocircuitos arteriovenosos (derivaciones tanto de la circulación pulmonar como de la general Cortocircuitos arteriovenosos + disminución de la viscocidad de la sangre por anemia incremento del GC Px con miocardiopatía alcohólica desarrollan ICC Alteración en el intercambio de gases pulmonares así como en la mecánica ventilatoria. Hiperventilación produce alcalósis respiratoria primaria Hipoxemia por cortocircuito de derecha a izq (hasta el 40% del GC) Cortocircuito se debe al incremento de comunicaciones arteriovenosas pulmonares como la desigualdad V/Q (relativo) La elevación del diafragma a causa de la ascitis disminuye los volumenes pulmonares, en especial la capacidad funcional residual y predispone a atelectasia.
- 40. Cantidades grandes de ascitis producen un defecto ventilatorio restrictivo que incrementa el trabajo de la respiración. Paracentésis debe efectuarse con cuidado ya que la extracción excesiva de líquido puede causar colapso circulatorio. MANIFESTACIONES RENALES Y EQUILIBRIO HÍDRICO Los cambios en el equilibrio de líquidos y electrolitos se manifiestan con ascitis, edema, alterac. Electrolíticas ó Sd hepatorrenal Mecanismos causantes de Ascitis: 1. Hipertensión Portal que aumenta la presión hidrostática y favorece la trasudación de líquido a/v del intestino 2. Hipoalbuminemia, que disminuye la presión oncótica del plasma y también favorece la trasudación de líquido 3. Escape de líquido linfático rico en proteínas de la sueprficie serosa hepática, secundario a distorsión y obstrucción de los conductos linfáticos en hígado. 4. Retención renal ávida de sodio 5. Teoria de Llenado Incompleto ó de Rebosamiento (Retención de Sodio): 6. Aunque los volumenes de plasma y líquido extracelular total que son medibles se encuentran incrementados , el “volumen efectivo del plasma” disminuye; la retención de sodio es secundaria a la hipovolemia relativa y al hiperaldosteronismo secundario La ascitis es producto de la trasudación secundaria a un volumen plasmático expandido. Px con ascitis ptan catecolaminas circulantes aumentadas por el aumento en el flujo simpático Aumento de la renina y de la angiotensina II circulantes Inestabilidad al péptido natriurético auricular circulante Px con cirrósis y ascitis ptan. Disminución de la perfusión renal, alteración de la hemodinámica intrarrenal, aumento en la resorción de sodio proximal y distal y deterioro en la depuración de agua libre. Hiponatremia dilucional Hipopotasemia por pérdidas urinarias excesivas de potasio por hiperaldosteronismo secundario ó por diuréticos SD HEPATORRENAL: Defecto renal funcional en px con cirrosis que suele presentarse después de hemorragias digestivas diuresis severa, infección o cirugía mayor. Se caracteriza por oliguria progresiva, retención ávida de sodio, azoemia, ascitis intratable y un índice de mortalidad elevado Tx de sosten que no tiene éxito a menos que se practique transplante de hígado.
- 41. Tx perioperatorio líquido moderado Evitar la diuresis perioperatoria excesiva Corregir las deficiencias agudas de líquidos intravascularcon coloides IV Diuresis de ascitis y de líquido de edema se logra a lo largo de varios díasDiuréticos de ASA cuando son ineficaces el reposo en cama, la restricción de sodio (< 2 gm NaCl/día 9 Y la espironolactona. Peso corporal diario para prevenir la pre la depresión de volumen intravascular durante la diuresis Px con ascitis + edema periférico no eliminar más de 1K/día durante la diurésis Px con ascitis no deben perder más de Kg/día Hiponatremia (Na sérico < 130 mEq/L) restricción de agua (1.5 L/día) Deficiencias de potasio requiere reemplazo en el preoperatorio Manitol IV profiláctico para prevenri la Insuf Renal MANIFESTAC DEL SNC La encefalopatía hepática se caracteriza por alteraciones del estado mental (asterixis , hiperreflexia o reflejo plantar invertido) y cambios electrocardiográficos característicos (actividad simétrica de onda lenta de alto voltaje) Elevación de la PIC Encefalopatía metabólica se relaciona con la cantidad de daño hepatocelular y con el grado de derivación de sangre portal hacia fuera del hígado y directo a la circulación general Causa de acumulación de sustancias (amoníaco, metabolitos de la metionina ( mercaptanos) ácidos grasos de cadena corta y fenol) originadas en el tubo digestivo pero metabolizadas por el hígado Aumento de las concentraciones de aminoácidos aromáticos y disminución de los aminoácidos de cadena ramificada en sangre, aumento de la permeabilidad de la barrera hematoencefálica y concentraciones anormales elevadas de ácido gamama aminobutírico en el encéfalo. Factores precipitantes de encefalopatía incluyen : 1. Hemorragia digestiva 2. Aumento de la ingestión de proteínas 3. Alcalosis 4. Hipopotasemia por vómito o diurésis 5. Infecciones 6. Empeoramiento de la función hepática
- 42. CLASIFICACION CARACTERÍSTICAS GRADO I Hábitos sueños alterados, perdida de la orientación espacial GRADO II Asterixis presente GRADO III Estupuroso, responde a estímulos nocivos GRADO Iv Sin respuesta Descerebrado o postura de descorticación Tx de encefalopatía Corregir causas precipitantes Lactulosa 30 – 50 ml c/8h ó Neomicina 500 mg c/6h PO para reducir la absorción intestinal de amoníaco VALORACIÓN PREOPERATORIA El anestesiólogo debe establecer farmacoterapia actual, ictericia previa y actual, antecedentes de tranfusiones sanguíneas y hemorragia digestiva, operaciones y anestesias anteriores. Ex Físico: exploración rutinaria de órganos y sistemas, revisión cuidadosa del aspecto del px y el grado de ascitis y encefalopatía. Pruebas sanguíneas ESTRICTAS: Hb,,HTO,Recuento Plaquetario, Bilirrubina sérica, Electrolitos séricos (Sodio y Potasio), Creatinina y Nitrógeno de Urea Sanguínea (BUN), Gases en Sangre arterial, Proteínas séricas, TP y TPT, TGO, TGP, FA, Deshidrogenasa de Hidroxibutirato (Isoenzima de la Deshidrogenasa Láctica que es más específica para la función hepática) Valorar la función del sistema de Coagulación y tratarla de manera apropiada antes de la intervención. En px con Ictericia Obstructiva, que muestran TP y TPT anormales, puede ser benéfico un ensayo de vitamina K (10 mg IM TID unos cuantos días); si no tiene éxito o no hay tiempo para corregir la hipocoagulación con vitamina K, debe considerarse el tx con plasma fresco congelado. Corregir Trombocitopenia de una hepatopatía avanzada antes de la operación por transfusión de concentrados de plaquetas. Las cifras de plaquetas debe ser de 100,000/mm3 en un adulto promedio. ES NECESARIO CORREGIR LA COAGULOPATÍA DIAGNOSTICADA ANTES DE LA ANESTESIA RAQUÍDEA O EPIDURAL PREMEDICACIÓN Suministrar todos los medicamentos indispensables para controlar el estado patológico tomando en cuenta la capacidad reducida del hígado para metabolizar fármacos. DEBE OMITIRSE LOS SEDANTES O DISMINUIR LA DOSIS Los px con hepatopatía avanzada pueden tener el estómago lleno, incluso si no han ingerido alimento o líquido durante varias horas, debido a HERNIA HIATAL, ASCITIS MASIVA Y DISMINUCIÓN DE LA MOTLIDAD GÁSTRICA E INTESTINAL.
- 43. Por esta razón la premedicación puede incluir un bloqueador del receptor de Histamina H2 (p. ej. Ranitidina), Metoclopramida y Citrato de Sodio. INDUCCIÓN Estómago lleno Inducción de secuencia rápida o intubación traqueal despierto Actividad de Succinilcolina es normal, a pesar de cierta disminución de la Colinesterasa plasmática. En personas con una reducción de la actividad de Colinesterasa, puede prolongarse más el efecto de la succinilcolina en comparación con sujetos normales. RMND se eligen para facilitar la intubación endotraqueal (p. ej D-tubocurarina) tienen una gran afinidad por la globulina gamma y aumenta el volumen de distribución de dichos fármacos; por tanto, la dosis necesaria para lograr relajación suele ser más alta que en personas normales. Hepatopatía avanzada también está incrementado el volumen de distribución del Pancuronio. El Pancuronio se une mal a las proteínas del plasma y no existe una correlación entre el volumen de distribución y la concentración plasmática de la globulina gamma. Atracurio tiene alto volumen de distribución por el incremento sustancial del líquido extracelular. El volumen de distribución del Vecuronio NO se altera de manera significativa, por tanto no cabe esperar un cambio importante de la dosis eficaz necesaria de este relajante muscular. Por lo que se debe ajustar el medicamento con un estimulador neural transcutáneo. MANTENIMIENTO Es posible hipoxia por una FIO2 o hipoventilación inadecuadas HIPOXIA ANÉMICA si no se asegura la capacidad de transporte de oxígeno de la sangre (HTO Adecuado) HIPOXIA CIRCULATORIA por trastornos hemodinámicos sistémicos (hipovolemia, hipotensión arterial, reducción del gasto cardíaco) o regionales (disminución del aporte hepático de sangre y oxígeno): es posible que la disminución del aporte de hepático de sangre y oxígeno sea consecutivo a alteraciones circulatorias sistémicas o por manipulaciones alrededor del hígado y asimismo de los efectos de muchas sustancias vasoconstrictoras endógenas (p. Ej., renina-angiotensina, catecolaminas, hormona antidiurética), y HIPOXIA CITOTÓXICA Por la interferencia de los anestésicos con el transporte de electrones a nivel celular puede ocasionar
- 44. Respuesta a los anestésicos impredecible Aumento de la sensibilidad del SNC al Tyopental, mientras px alcohólicos ptan tolerancia Resistencia a los RMND por el incremento del volumen de distribución de estos farmacos ionizados y por la expansión del compartimiento del LEC por lo que se requieren dosis mayores de impregnación. Sin embargo se requieren dosis menores de mantenimiento para farmacos que se eliminan por el hígado (pancuronio, rocuronio y vecuronio) Prolongación de la duración de acción de la succinilcolina por la disminución en la concentración de seudocolinesterasa En la cirrosis el flujo sanguíneo venoso portal está reducido y el hígado está dependiente de l riego de la arteria hepática Anestesia regional en px sin trombocitopenia ni cuagulopatía, cuidados en la hipotensión Anestesia General: Barbitúricos, Insoflurano en oxígeno o una mezcla de O2+ N2O Evitar el Halotano La vida media de los opiodes se encuentra prolongada lo cual aumenta la duración de la depresión respiratoria Cisatracurio el RMND preferido por su metabolismo no hepático Oxigenación previa e inducción de secuencia rápida con presión cricoidea en px con náuseas, vómitos , hemorragia GI alta y distensión abdominal antes de la cirugía por ascitis masiva Px inestables con hemorragia activa intubación con el px despierto o inducción de secuencia rápida con presión cricoidea con Ketamina (o Etomidato) y succinilcolina. EKG en 5 derivaciones en px que reciben vasopresina para detectar isquemia miocárdica por vasoconstricción coronaria SPO2 Gases sanguíneos arteriales para evaluar estado ácido básico Px con cortocircuitos intrapulmonares de derecha a izq no toleren el N2O y pueden requerir PPFE para tratar la desigualdad de la relación V/Q yla hipoxemia
- 45. Vigilancia de P/A Invasiva Intraarterial por los cambios de P/A por la hemorragia excesiva, desplazamiento de líquidos entre compartimientos y manipulacíones quirúrgicas PVC para valorar estado de volumen intravascular, para la prevención del Sd hepatorrenal Vigilancia del gasto urinario, debe considerarse la admón de Manitol o dosis bajas de Dopamina CONSIDERACIONES TRANSOPERATORIAS REEMPLAZO DE LÍQUIDOS Se prefieren líquidos coloides para evitar la sobrecarga de sodio y aumentar la presión oncótica El reemplazo de líquidos debe tomar en consideración: 1. Hemorragia excesiva (Por congestión venosa por hipertensión portal, lisis de adherencias de cirugías previas y cuagulopatías) 2. Desplazamiento de líquido (Proced. abdominales) 3. Evacuación de ascitis 4. Procedimientos quirúrgicos prolongados Transfundir por anemia y alteraciones de la coagulación Las transfusiones excesivas provocan intoxicación por citrato por su metabolismo alterado ENFERMEDAD SGTP SGOT FOSFATASAS ALCALINAS Hemólisis Viral NL o NL NL Colestática NL 2 veces 2 - 10 Alcohólico NL 1 - 5 1 – 3 Neoplasmas 1 - 2 1 - 2 1 – 3 Drogas Hepático 2 - 10 2 - 10 Nl Colestática NL NL 2 - 5