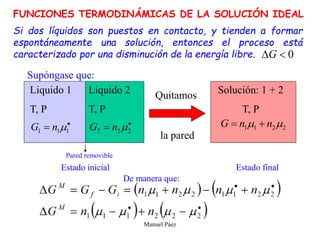
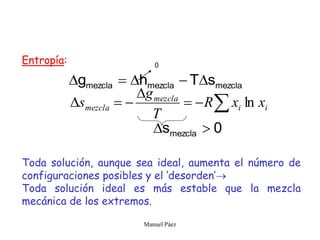
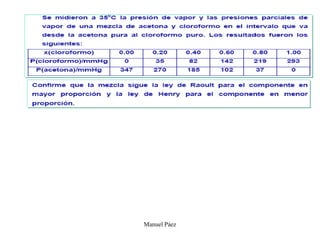
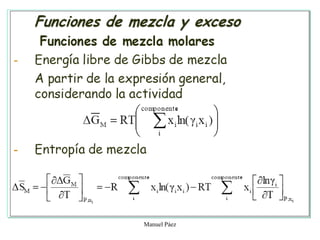
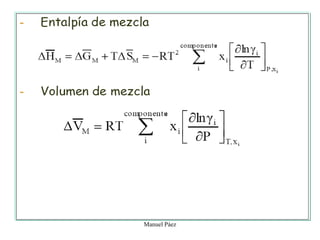
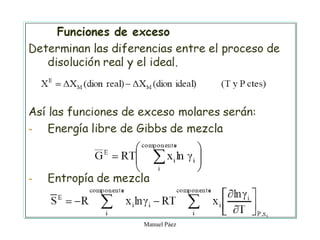
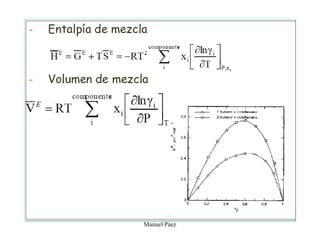
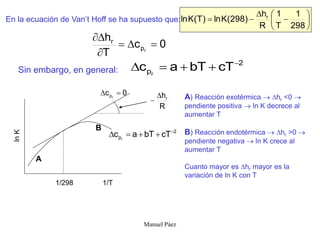
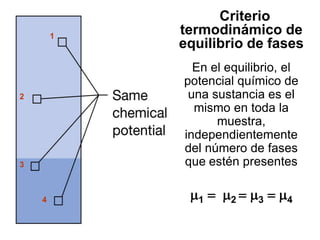
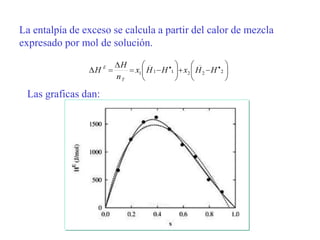
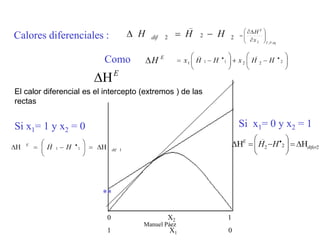
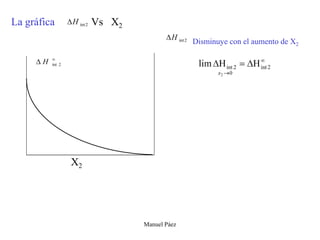
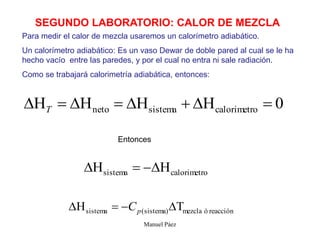
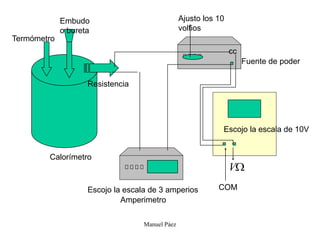
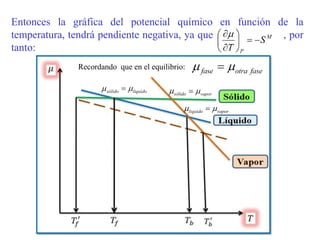
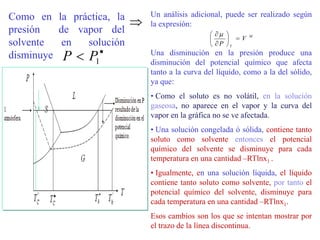
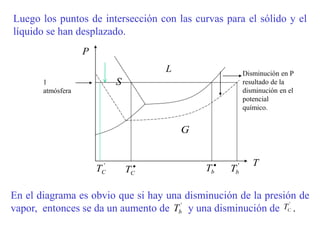
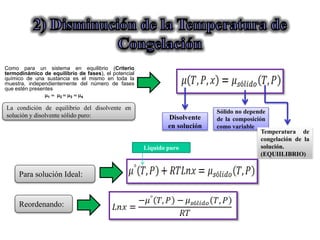
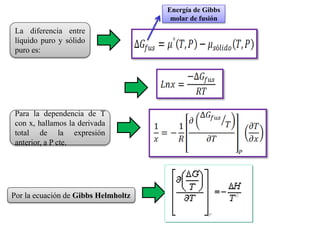
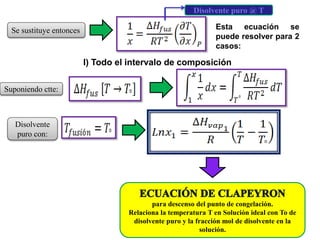
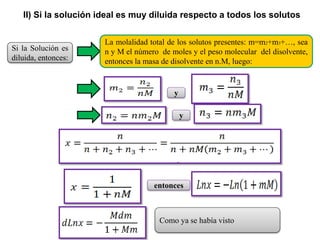
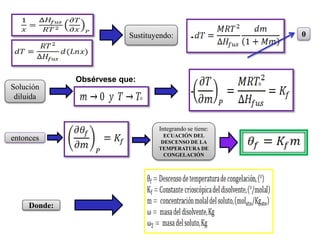
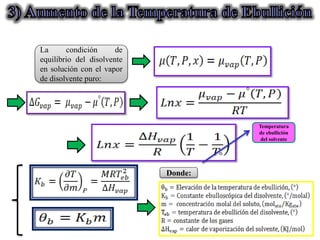
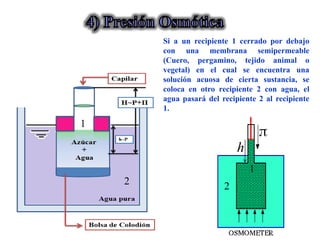
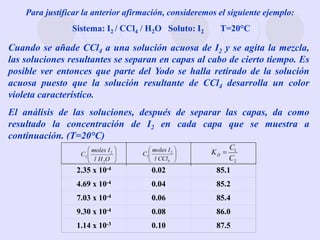
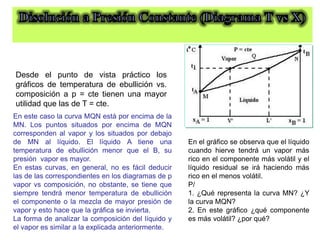
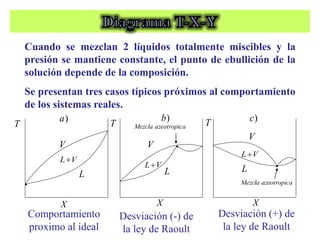
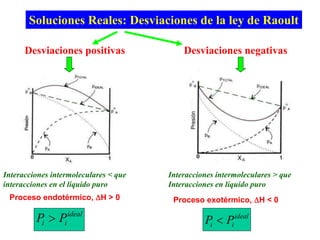
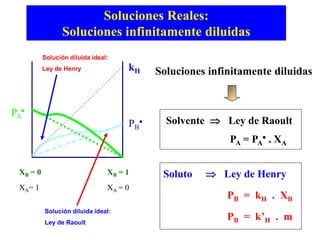
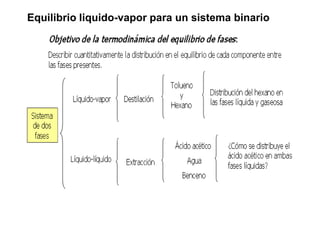
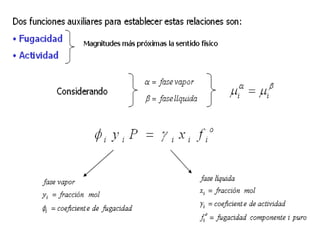
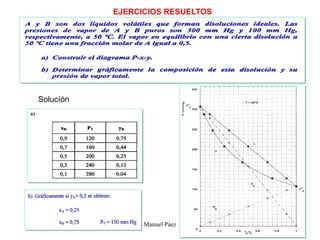
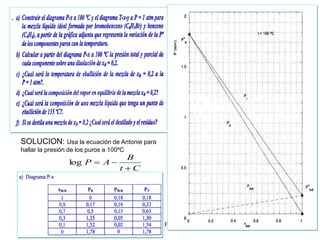
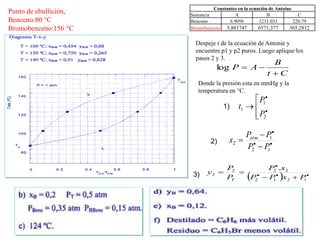
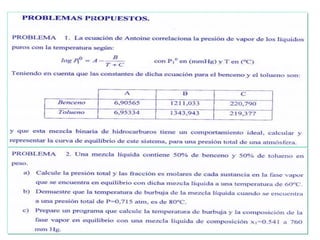
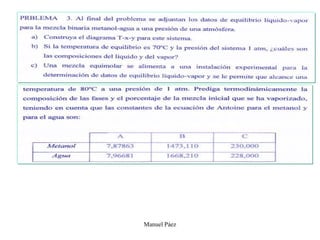
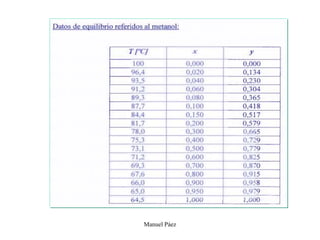
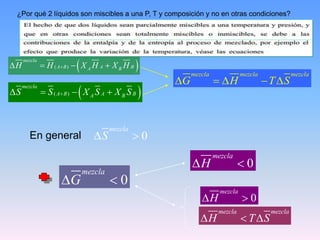
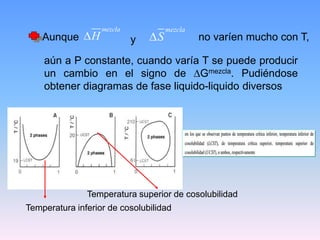
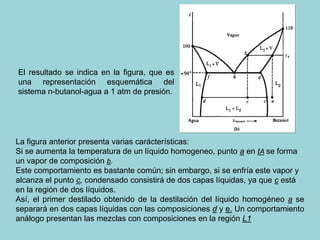
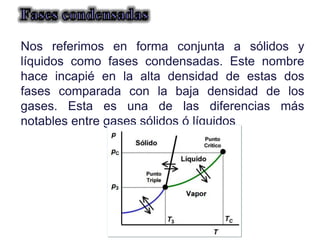
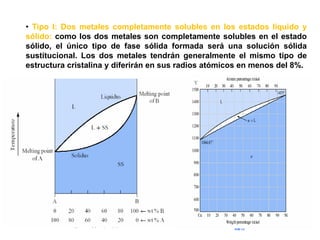
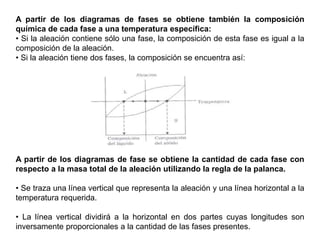
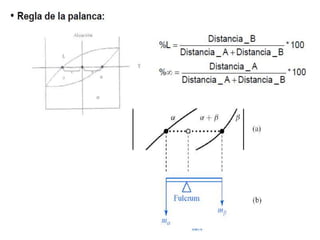
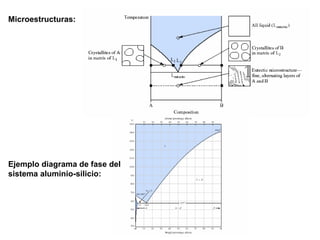
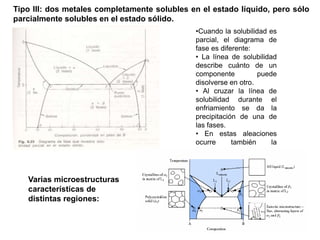
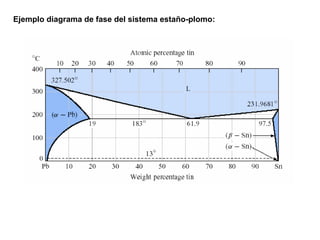
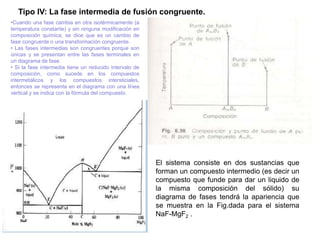
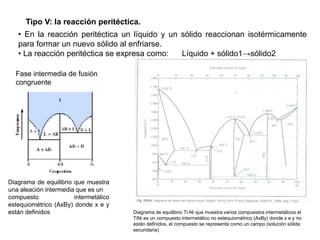
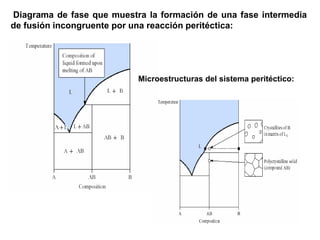
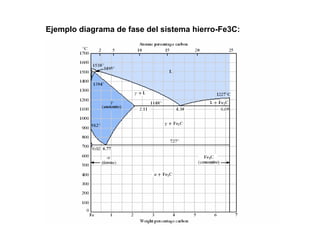
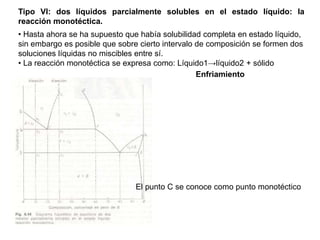
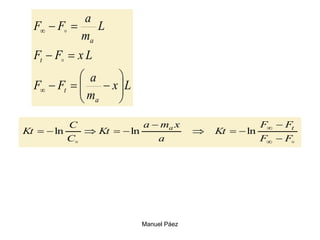
Este documento trata sobre la termodinámica de soluciones. Explica conceptos como soluciones ideales, potencial químico y propiedades parciales. También cubre temas como la energía de Gibbs, entalpía y volumen de mezclado para soluciones ideales, así como propiedades coligativas como la disminución de la presión de vapor y la temperatura de congelación. Finalmente, incluye ejemplos resueltos para demostrar relaciones entre las propiedades termodinámicas en sistemas de soluc








































































































![Considérese un soluto S que se asocia en la fase orgánica
dando Sn
donde n es el grado de asociación; y se
encuentra en forma monomérica en la fase acuosa .
De acuerdo con lo anterior en la fase ocurre el
equilibrio:
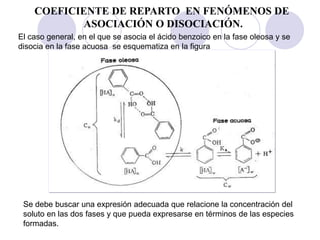
COEFICIENTE DE REPARTO EN FENÓMENOS DE
ASOCIACIÓN
La concentración [S] está relacionada con el
coeficiente de reparto del soluto en la fase orgánica
y la fase acuosa, mediante la expresión
Si se considera que la
asociación es
prácticamente completa, el
numerador de esta
expresión es igual a la [ ]
analitica](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-140-320.jpg)
![Eliminando la especie de ambas expresiones se obtiene:
De donde despejando
Se obtiene :
]
[
m
S
Lo que podría permitirnos usar el método gráfico
EJEMPLO
Conociendo gráficamente
Se evalúa y a partir de ella se obtiene
KD](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-141-320.jpg)







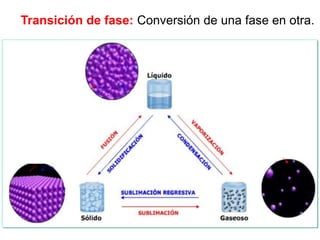
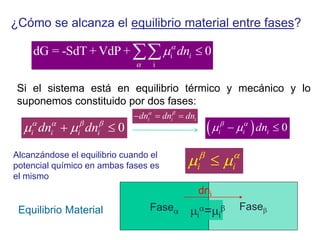
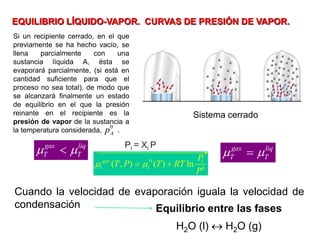
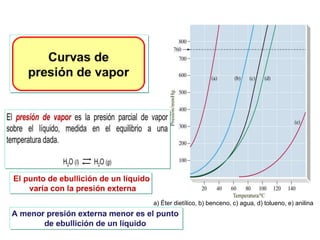




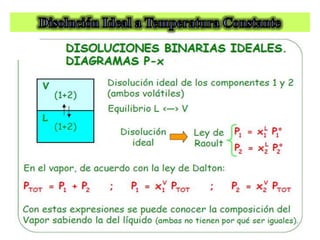
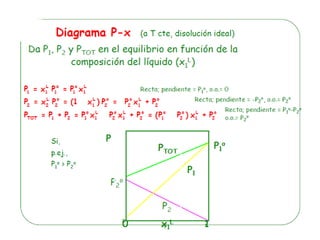
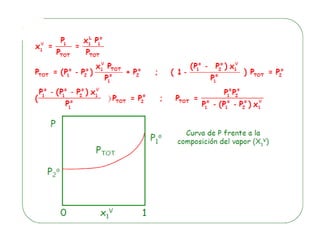

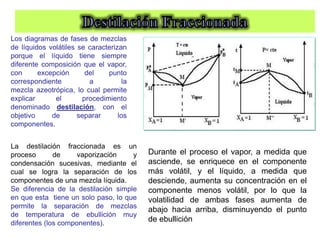













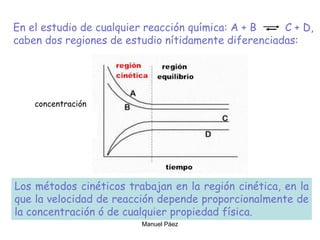
![Cuando en el sistema ocurren r reacciones químicas, el número de
variables independientes se reducen
F = C – P + 2 - r
Si además existen relaciones estequiométricas o de conservación de
la electroneutralidad, el número de variables intensivas
independientes es menor
F = C – P + 2 - r - a
Mezcla gaseosa : N2, H2 y NH3: C = 3, F = 1, GL = 3 - 1 +2 = 4
T , P, X1 y X2
NH3 con catalizador para establecer el equilibrio 2NH3 N2 +3 H2
C = 3 F = 1 r = 1 a = 1 [X(H2) = 3X(N2)]
GL = 3 – 1 + 2 – 1 - 1 = 2 T, P
r: Reacciones químicas que vinculan los componentes
a= otras relaciones estequiométricas o de presiones](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-199-320.jpg)
















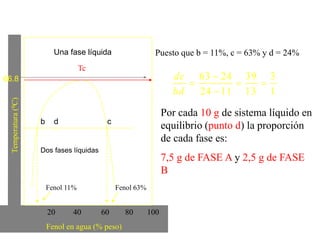



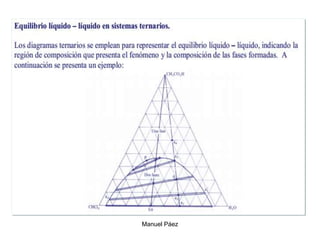

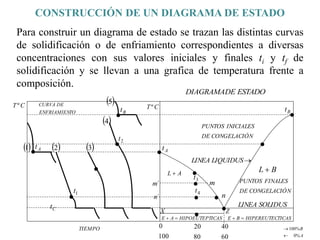




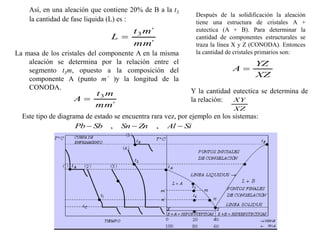



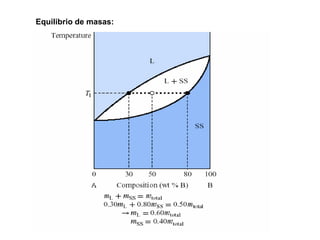
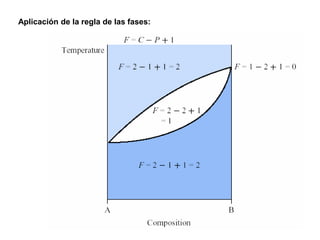
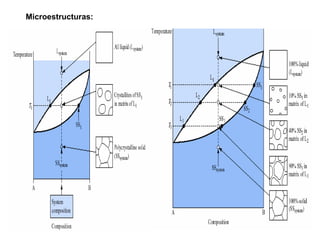

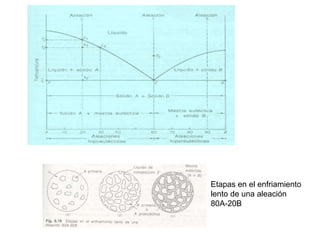


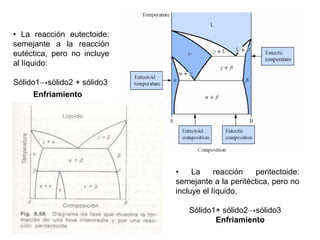








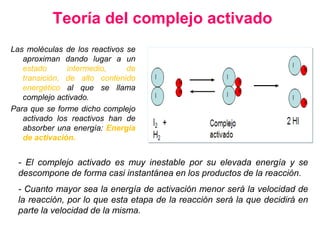
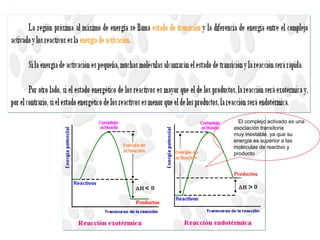
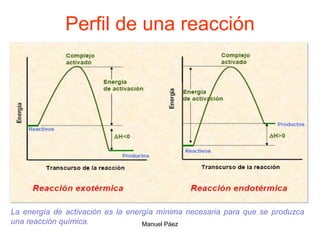
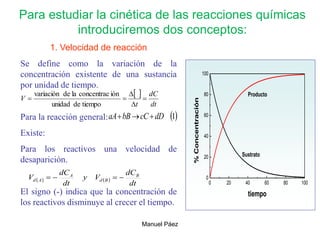
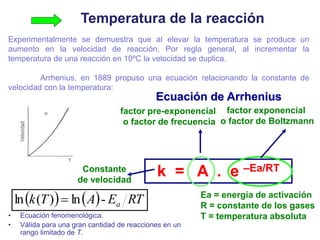
![2. Postulado Fundamental de la
Cinética
...
]
[
]
[
]
[
)
(
F
B
A
k
V T
k = constante de velocidad de reacción.
= orden de reacción respecto al componente A.
= orden de reacción respect al componente B.
+ +…+ = orden total de reacción.
a A + b B+... c C + d D
Únicamente en los casos en que la reacción transcurre en una sola etapa el orden
de la reacción para la sustancia dada coincide con el coeficiente estequiométrico.
Es importante notar que el orden la reacción es igual al coeficiente estequiométrico
para todas las reacciones que transcurren muy despacio en las condiciones
infinitamente cercanas al estado de equilibrio químico, independientemente de que
en las condiciones alejadas del equilibrio puedan pasar por una serie de etapas
intermedias.
En el caso general se puede admitir que la velocidad de la reacción química es
directamente proporcional a las concentraciones de las sustancias reaccionantes
con ciertos exponentes, denominados orden de reacción.](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-277-320.jpg)
![Manuel Páez
Comentarios sobre v =k[A][B][F]
• k no depende de la composición …
– pero depende de T, P, número de fases.
• Los órdenes pueden ser enteros o fraccionarios.
• Los órdenes no necesariamente son iguales a
los coeficientes estequiométricos.
• Los órdenes deben determinarse
experimentalmente.](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-278-320.jpg)

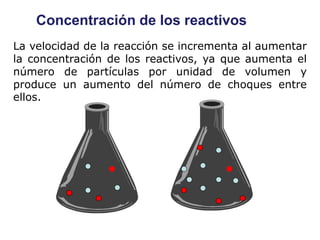


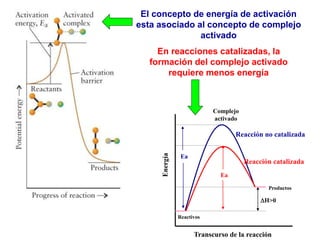

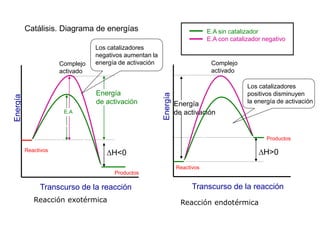


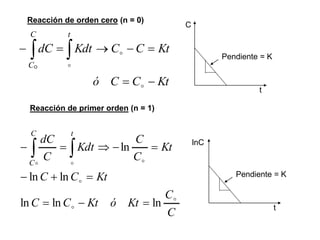
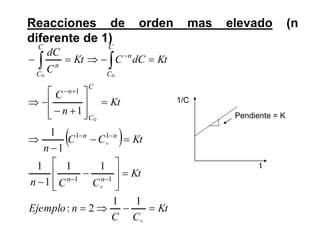




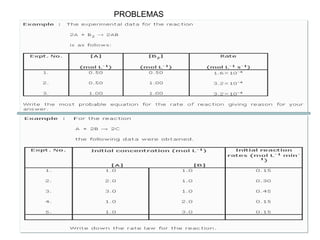






![II- Reacciones opuestas o reversibles
Reacciones que se producen en ambos sentidos y por lo general
conducen a un estado de equilibrio Vdirecta = Vinversa
A
B
Tiempo
[ ]
A B
k1
k-1
k1 [A] = k-1 [B]
K
k
k
A
B
1
1
]
[
]
[
constantes de
velocidad directa e
inversa
constante de
equilibrio
d(A)/dt = - k1 (A) + k-1 (B) = 0](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-307-320.jpg)







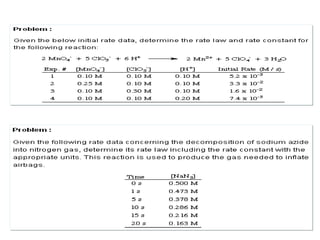
![Reacciones en las cuales el producto de una de las etapas
elementales es el reactante de la siguiente
A B C
k2 >>> k1 k2 <<< k1
k1 k2
A C
B
Tiempo
[ ]
A C
B
Tiempo
[ ]
III- Reacciones consecutivas o en series](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-315-320.jpg)









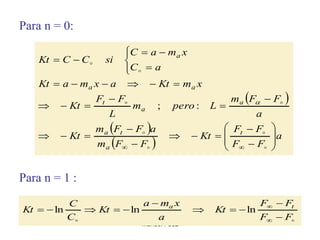









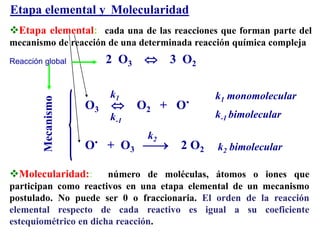
![Orden de reacción y molecularidad
ORDEN no es igual a MOLECULARIDAD
ORDEN
Magnitud empírica
determinada a partir de la
ley de velocidad
v = k [A]a [B]b
n = a + b
MOLECULARIDAD
Número de moléculas,
átomos o iones que
participan como reactivos
en una etapa elemental en
un mecanismo postulado
El orden de reacción coincide con la
molecularidad cuando la reacción se realiza en
una sola etapa, es decir, sin un mecanismo
involucrado](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-338-320.jpg)
![Manuel Páez
Velocidad de las reacciones
simples
En el caso de las reacciones simples, el orden
de reacción de cada especie coincide con su
molecularidad y velocidad viene dada por
M
P
M
B
A
A
A
A
D
C
B
A
B
A
2
]
][
][
[
]
[
]
][
[
]
[
2
M
B
A
k
v
A
k
v
B
A
k
v
A
k
v
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-339-320.jpg)
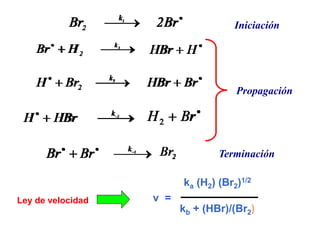
![Mecanismos de reacción
Reacciones elementales
Una de la reacciones (etapas) es la
mas lenta de todas las que conforman
el mecanismo
Etapa determinante de la
velocidad de la reacción
global
2A + 2B C + 2D
Mecanismo propuesto
2A I
I + B C + I2
I2 + B 2D
Reacción global
k1
k -1
k2
k3
Etapa mas lenta
V = k2 [I] [B]
Está constituido](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-340-320.jpg)
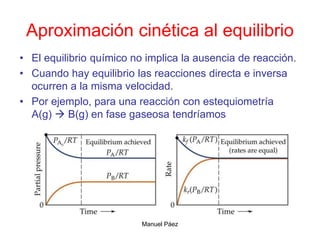
![Manuel Páez
Aproximación cinética al equilibrio
B
A
eq
eq B
k
A
k ]
[
]
[
]
[B
k
v
Inicialmente, la velocidad directa es grande y la inversa es nula.
Por este motivo la concentración de A disminuye y la de B aumenta.
Esto hace que la velocidad directa disminuya y la inversa aumente.
Eventualmente las velocidades se igualan.
c
eq
eq
K
k
k
A
B
]
[
]
[
]
[A
k
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-342-320.jpg)



![Manuel Páez
Ley de velocidad y mecanismo
Reacción
C
A
C
B
B
A
k
k
2
1
Mecanismo
]
[
]
[
]
[
]
[
]
[
]
[
]
[
2
2
1
1
B
k
dt
C
d
B
k
A
k
dt
B
d
A
k
dt
A
d
• La ley se puede deducir del mecanismo.
• Sólo si el mecanismo propuesto es correcto la ley predicha
coincide con el experimento.
• Clave: para las etapas simples los órdenes de reacción
coinciden con la molecularidad de cada reactivo.
Sist. de ec
diferenciales](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-347-320.jpg)
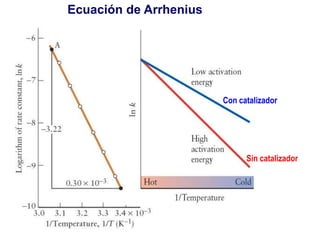
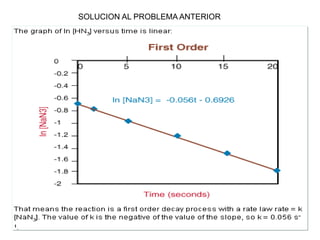
![Manuel Páez
Solución exacta
k1=1; k2=10
Tiempo
0.0 0.5 1.0 1.5 2.0
Concentraciones
0.0
0.2
0.4
0.6
0.8
1.0
1.2
[A]/[A]0
[B]/[A]0
[C]/[A]0
t
k
t
k
t
k
t
k
t
k
e
k
e
k
k
k
A
C
e
e
k
k
k
A
B
e
A
A
1
2
2
1
1
1
1
1
2
1
2
1
0
1
2
1
0
0
k1=10; k2=1
Tiempo
0.0 0.5 1.0 1.5 2.0
Concentraciones
0.0
0.2
0.4
0.6
0.8
1.0
1.2
[A]/[A]0
[B]/[A]0
[C]/[A]0](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-348-320.jpg)
![Manuel Páez
Soluciones aproximadas
Estado estacionario
• Se supone que la concentración de los
intermediarios (radicales libres) no cambia
con el tiempo
• Se utiliza la hipótesis anterior para expresar
la velocidad de aparición de un producto en
función de la concentración de especies
estables.
0
]
[
]
[
]
[
2
1
B
k
A
k
dt
B
d
]
[
]
[
]
[
1
2 A
k
B
k
dt
C
d
v
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-349-320.jpg)
![Manuel Páez
Ejemplos
]
[
]
[
1
]
][
[
2
2
2
2
2
1
2
2
Br
HBr
k
Br
H
k
v
HBr
Br
H
]
][
[
2 2
2
2
2 I
H
k
v
HI
I
H
2
2
3
2
2 ]
][
[
2
2 NO
O
k
v
SO
O
SO NO
]
][
][
[
+
]
][
[
2
2
2
2
2
2
2
2
1
2
2
2
2
H
I
O
H
k
I
O
H
k
v
I
O
H
H
I
O
H](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-350-320.jpg)
![Manuel Páez
Soluciones aproximadas
Hipótesis de pre-equilibrio I
Reacción C
A
C
B
B
A
k
k
k
2
1
1 /
Mecanismo
Sistema de ecuaciones diferenciales
]
[
]
[
]
[
]
[
]
[
]
[
]
[
]
[
]
[
2
2
1
1
1
1
B
k
dt
C
d
B
k
B
k
A
k
dt
B
d
B
k
A
k
dt
A
d Hay que resolver el
sistema de ecuaciones
diferenciales.
Otra opción es utilizar
la hipótesis de estado
estacionario para B.
Otra es asumir que el
primer paso está en
equilibrio.](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-351-320.jpg)
![Manuel Páez
Hipótesis de pre-equilibrio II
]
[
]
[
2 B
k
dt
C
d
1
1
1
]
[
]
[
k
k
K
A
B
Pero si hay cuasi-equilibrio en la 1ra etapa
]
[
]
[
]
[
]
[
1
1
2
1
2
A
k
k
k
dt
C
d
A
K
k
dt
C
d
Notar que la hipótesis de pre-equilibrio
es un caso particular de la hipótesis del
estado estacionario.](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-352-320.jpg)


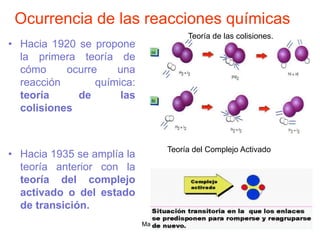
![Manuel Páez
Teoría de las colisiones de esferas
duras
reactivas
colisiones
de
fracción
tiempo
de
unidad
por
colisiones
de
numero
reacción
de
velocidad
]
][
[
8
2
1
2
2
B
A
T
k
d
N
Z B
AB
AB
B
A
B
A
m
m
m
m
2
B
A
AB
d
d
d
Factor de Boltzman
]
exp[ RT
E
f a
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-356-320.jpg)
![Manuel Páez
Fracción de colisiones efectivas
]
exp[ RT
E
f a
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-357-320.jpg)
![Manuel Páez
Teoría de las colisiones de esferas
duras
]
][
[
]
exp[
8
2
1
2
2
B
A
RT
E
T
k
d
N
v a
B
AB
T
f
Z
v AB
]
exp[
8
2
1
2
2
RT
E
T
k
d
N
k a
B
AB
col
¿Es consistente con Arrhenius?](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-358-320.jpg)


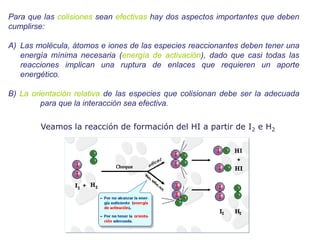
![Manuel Páez
La teoría del estado de transición
P
AB
B
A k
K
2
*
Reacción:
P
B
A
Eyring asume que hay un cuasi-equilibrio entre los reactivos y el complejo
activado y que luego este complejo activado se descompone para dar
productos
]
][
[
]
[
B
A
AB
K
]
[
2
AB
k
v ]
][
[
2 B
A
K
k
v
](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-361-320.jpg)
![Manuel Páez
La teoría del estado de
transición
RT
G
K
*
0
exp
]
][
[
exp
exp
*
0
*
0
2 B
A
k
v RT
H
R
S
h
T
k
k B
2
RT
H
R
S
K
*
0
*
0
exp
exp
]
][
[
exp
exp
*
0
*
0
B
A
h
T
k
v RT
H
R
S
B
kTST
¿Es consistente con Arrhenius?
¿Cuál es el sentido de H#
y S#?](https://image.slidesharecdn.com/fisicoquimicaii20231-230501165137-12d9c62e/85/Fisicoquimica-II-2023-1-ppt-362-320.jpg)