Descargado 458 veces



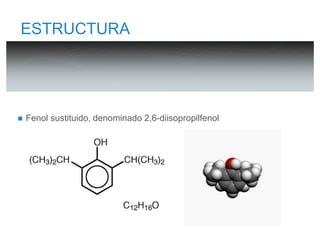






























![+
SNC
Principal efecto: Hipnosis
No actividad analgésica
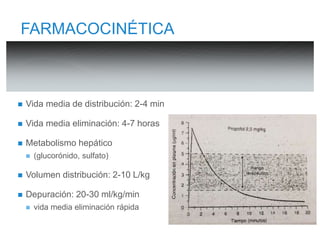
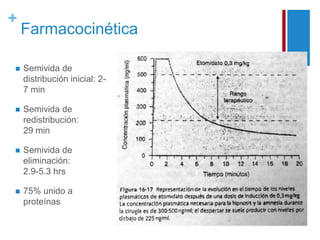
[ ] plasmática para mantenimiento 300-500 ng/ml
Sedación: 150-300 ng/ml
Despertar: 150-250 ng/ml
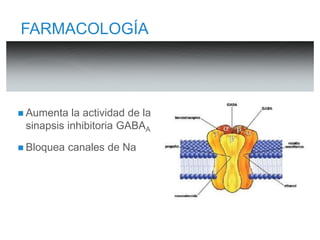
Mecanismo de acción similar al propofol,
subunidades B2 y B3 del receptor GABAA](https://image.slidesharecdn.com/inductores-141006202126-conversion-gate01/85/Inductores-Anestesicos-44-320.jpg)



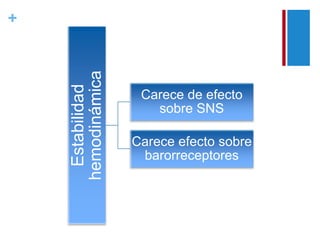
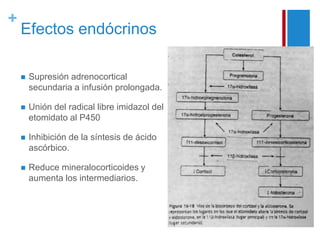




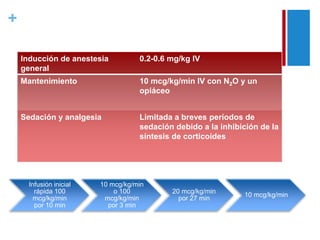
El documento describe las propiedades y usos del propofol y otros inductores de la anestesia general como la ketamina y el etomidato. El propofol es un sedante hipnótico altamente lipofílico que se usa comúnmente para la inducción y mantenimiento de la anestesia. La ketamina produce anestesia disociativa y una analgesia profunda sin depresión cardiovascular o respiratoria significativa. El etomidato proporciona estabilidad hemodinámica y mínima depresión respiratoria pero puede inhibir temporalmente la sí





























![ANESTESICOS INDUCTORES[2050].pdf](https://cdn.slidesharecdn.com/ss_thumbnails/anestesicosinductores2050-230424083733-7918181e-thumbnail.jpg?width=640&height=640&fit=bounds)





































