Descargado 167 veces
















![ Sans-Sabrafen, J, Besses, C, Vives, J.L. Hematología Clínica. (4ta
ed.). Madrid: ELSEVIER; 2001.
Pérez, J.C, Gómez, D. Hematología: La sangre y sus
enfermedades. (2da ed.). México: McGraw Hill; 2009.
Sefhes. 1. Sefhes. [Online]. Available from:
http://www.sefh.es/bibliotecavirtual/fhtomo2/CAP10.pdf [Accessed 5
January 2016].
Medynetcom. 1. Medynetcom. [Online]. Available from:
http://www.medynet.com/usuarios/jraguilar/Manual de urgencias y
Emergencias/leucemia.pdf [Accessed 5 January 2016].
Basesmedicinacl. 1. Basesmedicinacl. [Online]. Available from:
http://www.basesmedicina.cl/hematologia/15_5_leucemias/15_5_leuc
emias.pdf [Accessed 5 January 2016].](https://image.slidesharecdn.com/leucemiasagudas-160305074324/85/Leucemias-agudas-22-320.jpg)


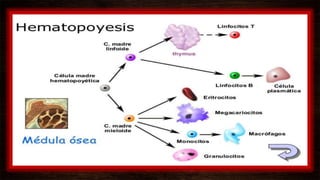
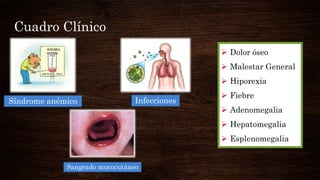
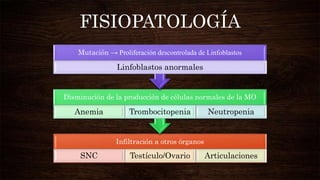
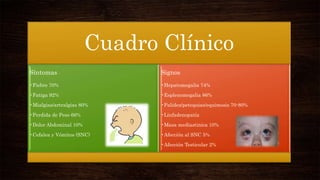
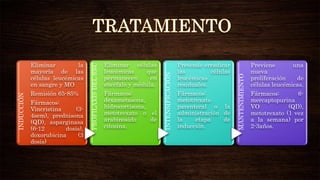
Las leucemias agudas se caracterizan por la proliferación anormal de células sanguíneas inmaduras. Presentan cuadros clínicos de anemia, sangrado y riesgo de infecciones. El diagnóstico requiere exámenes de sangre y médula ósea, e incluye identificar el tipo celular afectado. El tratamiento consiste en quimioterapia intensiva para inducir la remisión de la enfermedad.























































































