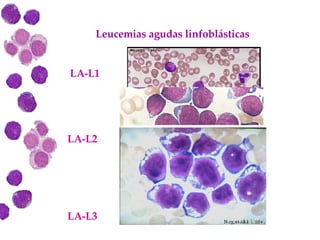
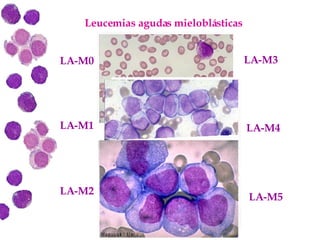
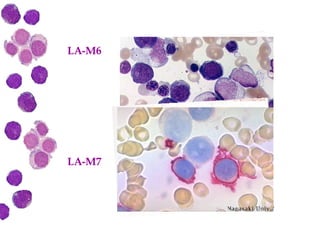
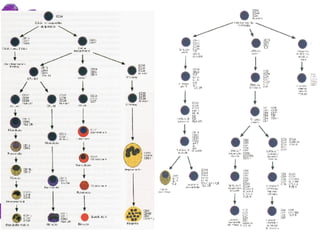
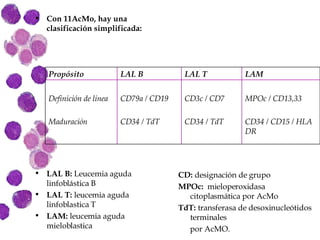
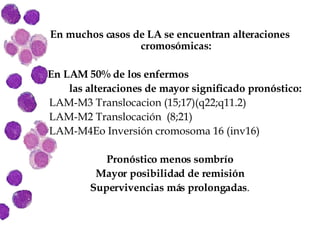
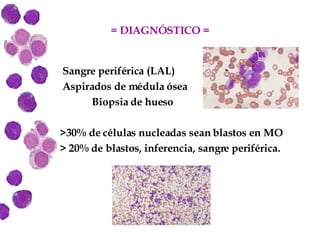
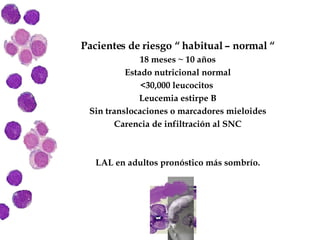
Las leucemias agudas son un grupo heterogéneo de enfermedades que suponen la proliferación descontrolada de células hematopoyéticas. Se clasifican en leucemias agudas mieloblásticas y leucemias agudas linfoblásticas según el tipo de célula afectada. El tratamiento incluye quimioterapia para inducir la remisión y prevenir recaídas, aunque el pronóstico depende de factores como la edad, el tipo de leucemia y las alteraciones cromosómic