Descargado 141 veces




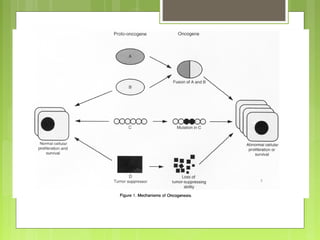

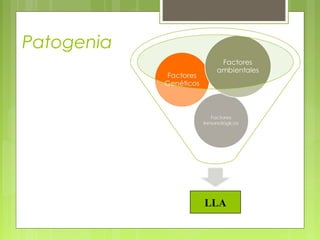
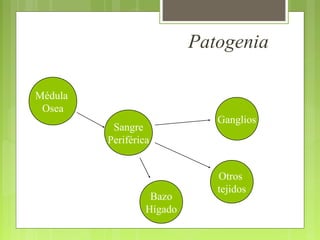

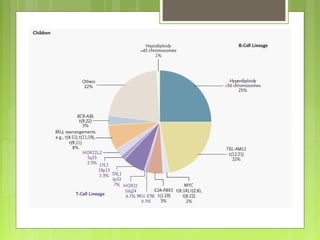



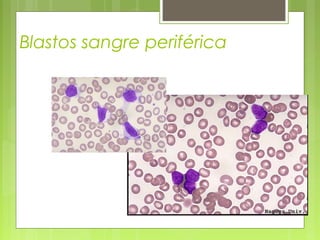


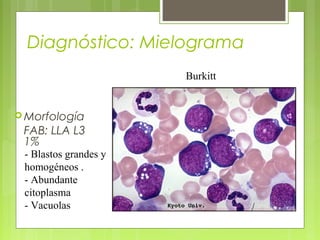


















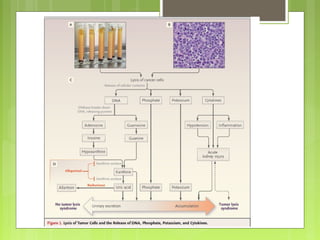





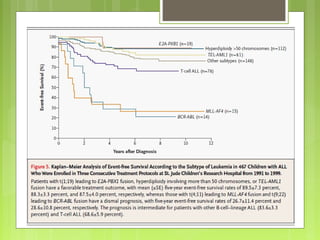





La leucemia linfoblástica aguda es una enfermedad causada por la proliferación anormal de células linfoides inmaduras en la médula ósea. Se caracteriza por presentar más del 25% de blastos linfoides en la médula ósea. Su tratamiento incluye quimioterapia de inducción, consolidación e intensificación, así como profilaxis y tratamiento del sistema nervioso central, con el objetivo de lograr la remisión completa de la enfermedad.























































































