
Recomendados
Recomendados
Más contenido relacionado
Similar a VASCULITIS.pptx
Similar a VASCULITIS.pptx (20)
Último
Último (20)
VASCULITIS.pptx
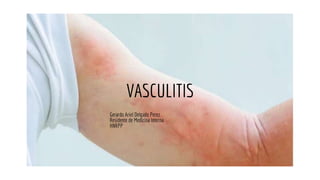
- 1. VASCULITIS Gerardo Ariel Delgado Perez Residente de Medicina Interna HNRPP
- 2. DEFINICIÓN Las vasculitis sistémicas son enfermedades incapacitantes heterogéneas caracterizadas por una inflamación crónica de los vasos sanguíneos que puede provocar destrucción de tejidos e insuficiencia orgánica Vasculitis sistémica: revisión de un año 2023 / M. Moretti et al
- 7. CLASIFICACIÓN Update of Systemic Vasculitides Nomenclature. International Chapel Hill Consensus Conference, 2012
- 8. CLASIFICACIÓN Update of Systemic Vasculitides Nomenclature. International Chapel Hill Consensus Conference, 2012
- 9. CLASIFICACIÓN clasificación de las vasculitis DCVAS (ACR/EULAR 2022)
- 10. VASCULITIS DE GRANDES VASOS ARTERITIS DE CÉLULAS GIGANTES La arteritis de células gigantes es una forma de vasculitis relativamente frecuente en los Estados Unidos y Europa. Es más usual en mujeres. La edad promedio al comienzo es de 70 años. Cerca del 40 al 60% de los pacientes con arteritis de células gigantes tienen síntomas de polimialgia reumática ARTERITIS DE TAKAYASU La arteritis de Takayasu (AT) es una rara vasculitis primaria y granulomatosa de grandes vasos de origen desconocido que afecta predominantemente a la aorta y sus ramas principales.
- 12. VASCULITIS DE GRANDES VASOS - CG AFECTA CARÓTIDAS AORTA TORÁCICA AORTA CERVICAL Y EXTRACRANEALES Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0
- 13. VASCULITIS DE GRANDES VASOS - CG EPIDEMIOLOGÍA - Frecuente en los Estados Unidos y Europa. Usual en mujeres. - 70 años, con un rango de entre 50 a > 90. Cerca del 40 al 60% de los pacientes con arteritis de células gigantes tienen síntomas de polimialgia reumática. - Por lo general, los vasos intracraneanos no están afectados. Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0
- 14. VASCULITIS DE GRANDES VASOS - CG SÍNTOMAS ACV, Cefaleas, alteraciones visuales, dolor a la presión en la arteria temporal y dolor en los músculos mandibulares durante la masticación. Fiebre, la pérdida de peso, el malestar general y la astenia. Aneurismas Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0
- 15. VASCULITIS DE GRANDES VASOS - CG DIAGNÓSTICO PCR VSG CLÍNICA BIOPSIA DE LA TEMPORAL HEMOGRAMA
- 16. VASCULITIS DE GRANDES VASOS - CG FISIOPATOLOGÍA MULTIFOCAL LOCALIZADA DISEMINADA Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0
- 17. VASCULITIS DE GRANDES VASOS - CG FISIOPATOLOGÍA ADVENTICIA Infiltrados de células mononucleares GRANULOMAS (CEL. T MACROFAGOS) LAS CÉLULAS GIGANTES CERCA A LA LÁMINA ELÁSTICA CAPA ÍNTIMA ENGROSADA CON ESTRECHEZ Y OCLUSIÓN
- 18. VASCULITIS DE GRANDES VASOS - CG Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0 Pacientes > 55 años y evidencia de laboratorio de inflamación sistémica: - Un nuevo tipo de cefalea - Un síntoma o signo nuevo compatible con isquemia de una arteria por encima del cuello - Dolor muscular mandibular durante la masticación - Dolor en la arteria temporal o el cuero cabelludo - Fiebre o anemia subagudas inexplicables - Es más probable si el paciente tiene también síntomas de polimialgia reumática.
- 19. VASCULITIS DE GRANDES VASOS - CG TRATAMIENTO TOXILIZUMAB CORTICOIDES ASPIRINA Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0
- 20. VASCULITIS DE GRANDES VASOS - CG - TTO Chrysidis S, Døhn UM, Terslev L, et al: Diagnostic accuracy of vascular ultrasound in patients with suspected giant cell arteritis (EUREKA): a prospective, multicentre, non-interventional, cohort study. The Lancet Rheumatology 3 (12) e865-e873, 2021. doi.org/10.1016/S2665-9913(21)00246-0 1. Dosis inicial de prednisona de 40 a 60 mg por vía oral 1 vez al día durante 4 semanas, seguida de una reducción gradual. 2. Alteraciones visuales, dosis inicial de metilprednisolona IV, 500 a 1.000 mg 1 vez al día durante 3 a 5 días para prevenir una mayor pérdida de visión. 3. Disminuirse en forma gradual, desde alrededor de 60 mg/día, de acuerdo con la respuesta del paciente, de a 5-10 mg por día cada semana hasta 40 mg por día, de a 2,5-5 mg al día cada semana hasta 10 a 20 mg al día; luego se reduce más la dosis hasta que se suspende el fármaco. 4. La mayoría de los pacientes requieren por lo menos 2 años de tratamiento con corticoides. 5. Tratamientos alternativos. Tocilizumab, un antagonista del receptor de IL-6, debe considerarse cuando se inicia el tratamiento. 6. Los pacientes de edad avanzada que toman prednisona durante largos períodos deben recibir un fármaco antirresortivo para aumentar la masa ósea y para prevenir osteoporosis. 7. La aspirina en dosis bajas (81 a 100 mg por vía oral 1 vez al día)
- 21. ARTERITIS DE TAKAYASU EPIDEMIOLOGIA La etiología se desconoce. Puede deberse a mecanismos de inmunidad mediada por células. En Norteamérica, la incidencia anual se estima en 2,6 casos/millón. 05 Edad de comienzo típica es entre los 15 y 30 años. 04 La relación mujeres:hombres es de 8:1 03 Común en Asia, pero ocurre en todo el mundo. 02 Es rara Predomina en mujeres jóvenes. 01 Aproximadamente un 50% de los pacientes refieren síntomas generales como fiebre, malestar general, sudoración nocturna, pérdida de peso, fatiga y/o artralgias
- 22. ARTERITIS DE TAKAYASU La arteritis de Takayasu afecta sobre todo a las grandes arterias elásticas. Los afectados con mayor frecuencia son ● Arterias braquiocefálica y subclavia ● Aorta (principalmente la aorta ascendente y el arco) ● Arterias carótidas comunes ● Arterias renales AFECTA AORTA ARTERIAS PULMONARES
- 23. ARTERITIS DE TAKAYASU CLASIFICACION
- 24. ARTERITIS DE TAKAYASU FISIOPATOLOGIA Infiltrado mononuclear en la adventicia con manguitos perivasculares en los vasa vasorum Infiltración mononuclear intensa de la media, a veces acompañada por cambios granulomatosos, Células gigantes y necrosis en placas de la media. Los cambios morfológicos pueden ser indistinguibles de los de la arteritis de células gigantes.
- 25. ARTERITIS DE TAKAYASU - CLÍNICA - El pulso arterial en brazos y piernas puede estar disminuido y ser asimétrico. - Soplos sobre las arterias subclavias, braquiales, carótidas, aorta abdominal y femorales. - Mareos, síncope, hipotensión ortostática, cefaleas, alteraciones visuales transitorias, accidentes isquémicos transitorios o accidente cerebrovascular. - Angina pectoral o infarto de miocardio. - La obstrucción de la aorta torácica descendente produce signos de coartación de aorta. - Se ven afectadas las arterias pulmonares, lo que a veces causa hipertensión pulmonar.
- 26. ARTERITIS DE TAKAYASU - DIAGNÓSTICO Arteriografía aórtica o angiografía por resonancia magnética. Estenosis, oclusión, irregularidades en la luz arterial, dilatación postestenótica, arterias colaterales alrededor de los vasos obstruidos y aneurismas. Anemia de enfermedad crónica, elevada concentración de plaquetas, en ocasiones recuento leucocitario elevado, y eritrosedimentación y proteína C reactiva elevadas.
- 27. ARTERITIS DE TAKAYASU - CUADRO CLÍNICO Isquemia de los órganos irrigados por la aorta o sus ramas, en especial en mujeres jóvenes. Soplo arterial y discrepancias en el pulso o la tensión arterial entre el lado derecho y el izquierdo o entre los miembros superiores e inferiores también sugieren el diagnóstico.
- 28. ARTERITIS DE TAKAYASU TRATAMIENTO 1. Corticoides y otros inmunosupresores 2. Cirugía de derivación. - Prednisona 1 mg/kg por vía oral 1 vez al día durante 1 a 3 meses; Cerca de la mitad de los pacientes tienen recidivas al disminuir o suspender el fármaco, a pesar de la respuesta inicial. - Metotrexato es de 0,3 mg/kg 1 vez por semana, y se aumenta hasta 25 mg/semana. - Aspirina 325 mg por vía oral 1 vez al día) 1. Mekinian A, Neel A, Sibilia J, et al: Efficacy and tolerance of infliximab in refractory Takayasu arteritis: French multicentre study. Rheumatology 51:882–886, 2012. doi: 10.1093/rheumatology/ker380 2. Mekinian A, Comarmond C, Resche-Rigon A, et al: Efficacy of biological-targeted treatments in Takayasu arteritis: Multicenter, retrospective study of 49 patients. Circulation 132:1693–1700, 2015. doi: 10.1161/CIRCULATIONAHA.114.014321
- 29. VASCULITIS DE VASOS MEDIANOS POLIARTERITIS NODOSA
- 30. VASCULITIS DE VASOS MEDIANOS - PAN Vasculitis necrosante sistémica que afecta las arterias musculares medianas y en ocasiones las arterias musculares pequeñas, y produce isquemia tisular secundaria. Riñones, la piel, los músculos, los nervios periféricos y el tracto digestivo No suele perjudicar los pulmones. El diagnóstico requiere biopsia o arteriografía. El tratamiento con corticoides e inmunosupresores suele ser efectivo.
- 31. VASCULITIS DE VASOS MEDIANOS - PAN - EPIDEMIOLOGÍA La poliarteritis nudosa (PAN) es rara (cerca de 2 a 33 casos/millón). Afecta sobre todo a adultos de edad mediana, y la incidencia aumenta con la edad, con un pico en la sexta década de la vida.
- 32. VASCULITIS DE VASOS MEDIANOS - PAN - ETIOLOGÍA La mayoría de los casos son idiopáticos. Un 20% de los pacientes tiene hepatitis B o C. La causa se desconoce, aunque participan mecanismos inmunitarios. Una causa pueden ser los fármacos. En pacientes con cierto tipo de linfomas y leucemias, artritis reumatoide o síndrome de Sjögren, puede aparecer una vasculitis sistémica similar a la poliarteritis nudosa (a veces llamada poliarteritis nudosa secundaria).
- 33. FISIOPATOLOGÍA - POLIARTERITIS NODOSA Inflamación transmural necrosante segmentaria de las arterias musculares. Más frecuente en los puntos de bifurcación. No afecta las vénulas o venas postcapilares. Las lesiones iniciales contienen leucocitos polimorfonucleares y en ocasiones eosinófilos; las lesiones más tardías contienen linfocitos y células plasmáticas. No se observa inflamación granulomatosa. La proliferación de la íntima con trombosis y oclusión secundaria produce infarto de órganos y tejidos. La debilidad de la pared arterial muscular puede causar pequeños aneurismas y disección arterial. La curación puede producir fibrosis nodular de la adventicia.
- 34. SINTOMATOLOGIA - POLIARTERITIS NODOSA Fiebre, cansancio, sudores nocturnos, falta de apetito, pérdida de peso y debilidad generalizada. Son frecuentes las mialgias en áreas de miositis isquémica focal y las artralgias. Los músculos afectados son sensibles a la palpación y pueden presentar debilidad. Puede haber artritis. Sistema nervioso periférico: mononeuritis múltiple con signos de afectación motora y sensitiva de los nervios peroneo, mediano o cubital. A medida que se ven afectados otras ramas nerviosas, el paciente puede presentar polineuropatía simétrica distal. Sistema nervioso central: cefalea y convulsiones. accidente cerebrovascular isquémico y hemorragia cerebral, en algunos casos causado por hipertensión. Renales: si se ven afectadas las arterias pequeñas y medianas de los riñones, el paciente puede tener hipertensión, oliguria, uremia y un sedimento urinario inespecífico con hematuria, proteinuria y sin cilindros celulares. La hipertensión puede agravarse rápidamente. La rotura de aneurismas de la arteria renal puede causar hematomas perirrenales. En casos graves, puede haber múltiples infartos renales con dolor lumbar y hematuria macroscópica. Puede haber insuficiencia renal por isquemia e infarto renal.
- 35. SINTOMATOLOGIA - POLIARTERITIS NODOSA Digestivas: la vasculitis del hígado o la vesícula biliar Cardíacas: miocardiopatía isquémica o hipertensiva. Cutáneas: puede haber livedo reticularis, Los nódulos en la PAN se asemejan al eritema nudoso, pero a diferencia de los nódulos del eritema nudoso, los de la PAN pueden ulcerarse, y presentan vasculitis necrosante visible en la biopsia, dentro de las paredes de las arterias medianas, por lo general en la dermis profunda y en el tejido subcutáneo. Genitales: puede haber orquitis con dolor y sensibilidad testicular.
- 36. diagnostico - POLIARTERITIS NODOSA - Fiebre inexplicable, artralgia, nódulos subcutáneos, úlceras en la piel, dolor en el abdomen o extremidades, caída del pie o de la muñeca de reciente comienzo, o hipertensión de rápido desarrollo, artralgia, nódulos subcutáneos, úlceras en la piel, dolor en el abdomen o extremidades, caída del pie o de la muñeca de reciente comienzo, o hipertensión de rápido desarrollo. - Arteriografía si los tejidos con afectación clínica no pueden biopsiarse - La angiografía por resonancia magnética no es el mejor estudio para diagnóstico. La biopsia, se prefieren las muestras de tejido subcutáneo, del nervio sural y de músculos en los que se sospecha afectación, a las muestras de riñón o hígado. Electromiograma y estudios de conducción nerviosa para seleccionar el sitio de biopsia muscular o nerviosa. Laboratorio son inespecíficos. Se debe hacer estudio de hepatitis B y C. Debido a que la glomerulonefritis no es una característica de la PAN, los cilindros eritrocíticos no son evidentes en el análisis de orina.
- 37. PRONOSTICO DE POLIARTERITIS NUDOSA Sin tratamiento, la supervivencia a 5 años es < 15%. Con tratamiento, la supervivencia a 5 años es > 80%, aunque puede ser más baja en pacientes con hepatitis B. Los siguientes hallazgos se asocian con un mal pronóstico: ● Insuficiencia renal ● Compromiso gastrointestinal ● Afectación neurológica
- 38. TRATAMIENTO DE POLIARTERITIS NUDOSA ● Corticoides solos o en combinación con ciclofosfamida, metotrexato o azatioprina, según la gravedad de la enfermedad ● Tratamiento de la hepatitis B cuando esté indicado El tratamiento de la poliarteritis nudosa depende de la gravedad de la enfermedad. En enfermedad grave con manifestaciones neurológicas, renales, digestivas o cardíacas, se utiliza ciclofosfamida más corticoides. En enfermedad moderada, pueden utilizarse corticoides más metotrexato o azatioprina. La hipertensión debe tratarse en forma agresiva.
- 39. Poliarteritis nudosa relacionada con hepatitis El tratamiento está dirigido a suprimir rápidamente la inflamación y la terapia antiviral. Se utiliza un curso breve de corticoides durante algunas semanas. A veces se realizan plasmaféresis hasta que el antígeno e de la hepatitis B (HBeAg) es reemplazado por anticuerpos contra el antígeno e de la hepatitis B (anti-HBe) o hasta que se mantenga la recuperación clínica durante 2 a 3 meses. Si bien no se ha comprobado que este enfoque mejore la supervivencia, comparado con el tratamiento inmunosupresor solamente, puede reducir el riesgo de complicaciones de hepatitis B a largo plazo y suprimir los efectos colaterales del tratamiento prolongado con corticoides e inmunosupresores. El tratamiento con corticosteroides, a veces con inmunosupresores citotóxicos (sobre todo ciclofosfamida), fue efectivo a corto plazo pero no previno las recaídas y las complicaciones (p. ej., hepatitis crónica, cirrosis) debidas a la persistencia del virus hepatitis B. La
- 40. VASCULITIS DE VASO PEQUEÑO VASCULITIS ASOCIADAS AL ANCA VASCULITIS POR INMUNOCOMPLEJO - ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR - VASCULITIS CRIOGLOBULINEMICA - Vasculitis asociada a inmunoglobulina A (IgAV)
- 41. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR Es una vasculitis fulminante de pequeño vaso poco frecuente Afecta al lecho capilar de los riñones y los pulmones y que se caracteriza por la presencia de anticuerpos anti-membrana basal glomerular (MBG) En su forma completa, de anticuerpos anti-membrana basal alveolar (MBA). En consecuencia (nefritis anti-MBG) o como síndrome pulmonar-renal con hemorragia pulmonar grave.
- 42. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR EPIDEMIOLOGÍA Incidencia 0,6-1,8 casos por millón por año en poblaciones asiáticas y caucásicas europeas. Aparentemente afecta a hombres y mujeres por igual. Responsable del 1-5% de todos los casos de glomerulonefritis y es la causa de hasta el 10-15% de los casos de glomerulonefritis rápidamente progresivas con semilunas.
- 43. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR SIGNOS Y SÍNTOMAS Es bimodal Principalmente en la tercera y séptima décadas de la vida. El 80-90%) glomerulonefritis rápidamente progresiva, el 40 a 60% experimentará hemorragia pulmonar concurrente, y una minoría puede presentar enfermedad pulmonar aislada. La afección pulmonar pueden incluir tos, dificultad respiratoria, hemoptisis, dolor torácico e hipoxia anemia y en casos más graves, puede producirse insuficiencia respiratoria mortal, hemorragia pulmonar grave.
- 44. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR ETIOLOGÍA Afecta a los capilares glomerulares, a los capilares pulmonares o a ambos, frente al dominio no-colagenoso 1 de la cadena alfa-3 del colágeno tipo IV (alfa3(IV)NC1). Un tercio de los pacientes también desarrollará anticuerpos anticitoplasma de neutrófilo (ANCA). Las células T autorreactivas pueden desempeñar un papel en su patogenia y se ha descrito una fuerte correlación con HLA-DRB1*1501 y DRB1*1502 en poblaciones caucásicas.
- 45. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR DIAGNÓSTICO Detección de anticuerpos anti-MBG en suero o depositados en tejido, junto con características patológicas de GN con semilunas, con o sin evidencia de hemorragia alveolar. El examen histológico de la biopsia puede mostrar una extensa formación de semilunas (proliferación de células extracapilares), destrucción de MBG, inflamación intersticial y necrosis tisular.
- 46. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR TRATAMIENTO Plasmaféresis combinada con altas dosis de glucocorticoides y ciclofosfamida. Rituximab, ya sea como adyuvante de la terapia estándar o como sustituto de la ciclofosfamida. Casos graves la diálisis. El trasplante renal está desaconsejado siempre que se detecten autoanticuerpos circulantes. La remisión sostenida en ausencia de signos clínicos de recurrencia debe confirmarse cada 6 meses.
- 47. ENFERMEDAD ANTIMEMBRANA BASAL GLOMERULAR PRONÓSTICO Sin tratamiento, el pronóstico de la enfermedad anti-MBG es desfavorable. La hemorragia alveolar es la principal causa de muerte prematura. La gravedad y el pronóstico de la enfermedad renal se correlacionan con los niveles circulantes de anticuerpos anti-MBG.
- 48. VASCULITIS CRIOGLOBULINÉMICA Poco frecuente mediada por inmunocomplejos circulantes crioprecipitables en suero. Se manifiesta clínicamente por la clásica triada de púrpura, debilidad y artralgia.
- 49. VASCULITIS CRIOGLOBULINEMICA Epidemiología Prevalencia es aún desconocida. Común en el sur que en el norte de Europa o en Norteamérica. Prevalencia de la crioglobulinemia mixta ''esencial'' es de aproximadamente 1:100.000 (con una proporción mujer-hombre de 3:1),
- 50. VASCULITIS CRIOGLOBULINEMICA SIGNOS Y SINTOMAS Afectación variable de los órganos, lesiones cutáneas (púrpura ortostática, úlceras), hepatitis crónica, glomerulonefritis membranoproliferativa, neuropatía periférica, vasculitis difusa, afectación pulmonar intersticial y trastornos endocrinos. Algunos pacientes pueden desarrollar tumores hepáticos y linfáticos malignos.
- 51. VASCULITIS CRIOGLOBULINEMICA TIPOS Tipo II, los inmunocomplejos crioprecipitables se componen de IgM monoclonal, los autoanticuerpos, y de IgG policlonal, los autoantígenos Tipo III, los inmunocomplejos están compuestos de IgM oligo-/policlonal y de IgG policlonal. Tipo I están implicadas inmunoglobulinas monoclonales de un único isotipo y es una enfermedad distinta.
- 52. VASCULITIS CRIOGLOBULINEMICA ETIOLOGIA Se ha sugerido que la infección por el virus de la hepatitis C (VHC) juega un papel causal, junto con la contribución de factores genéticos y/o ambientales. El virus de la inmunodeficiencia humana (VIH) o el síndrome de Sjögren primario.
- 53. VASCULITIS CRIOGLOBULINEMICA DIAGNOSTICO Las crioglobulinas mixtas circulantes Bajos niveles del componente 4 del complemento Púrpura cutánea ortostática Patología vasculitis leucocitoclástica que afecta a los vasos de medio calibre o, más habitualmente, de pequeño calibre, fácilmente detectables en una biopsia de piel.
- 54. VASCULITIS CRIOGLOBULINEMICA TRATAMIENTO Primera línea terapia antiinfecciosa (antivirales). Trastorno linfoproliferativo o autoinmune terapia específica para la enfermedad. El uso de inmunomoduladores, inmunosupresores, corticoides y/o plasmaféresis, solos o en combinación/secuencia con antivirales, debe adaptarse a cada paciente en función de la progresión y la gravedad. El anti-CD20 (rituximab) complicaciones (nefropatía, neuropatía, vasculitis grave, etc.).
- 55. VASCULITIS CRIOGLOBULINEMICA PRONOSTICO La probabilidad de prevenir la persistencia y la recurrencia con el tratamiento temprano y la erradicación del VHC. El tratamiento temprano puede evitar la progresión del daño orgánico. El pronóstico global es desfavorable en pacientes con enfermedad renal, insuficiencia hepática, enfermedad linfoproliferativa y neoplasias. Tipo II y tipo III) predisposición al desarrollo de linfoma maligno de células B u otras neoplasias.
- 56. Vasculitis asociada a inmunoglobulina A (IgAV) (Púrpura de Schönlein-Henoch) - Afecta sobre todo a los pequeños vasos. - Frecuente en niños. - Púrpura palpable, artralgias, signos y síntomas digestivos y glomerulonefritis. - El diagnóstico es clínico en niños, pero en el adulto requiere biopsia. - Esta enfermedad suele ser autolimitada en niños y crónica en adultos. - Los corticoides alivian las artralgias y los síntomas digestivos, pero no alteran el curso de la enfermedad. - La glomerulonefritis progresiva puede requerir altas dosis de corticoides y ciclofosfamida.
- 57. FISIOPATOLOGÍA Inmunocomplejos que contienen IgA en los pequeños vasos de la piel y otros sitios. Los antígenos que pueden provocar esta enfermedad Virus, infección estreptocócica, fármacos, alimentos, picaduras de insectos y vacunas. La glomerulonefritis focal proliferativa segmentaria es típica, pero suele ser leve.
- 58. Signos y síntomas de la vasculitis asociada a inmunoglobulina A (IgAV) - Erupción purpúrica palpable en pies, piernas y en ocasiones en el tronco y los brazos. - Áreas de urticaria que se vuelven palpables y que a veces experimentan hemorragia y confluyen. - Fiebre y poliartralgia con dolor periarticular e hinchazón de tobillos, rodillas, caderas, muñecas y codos. - Síntomas digestivos, que incluyen dolor abdominal cólico, dolor a la palpación en el abdomen y melena. Suelen remitir al cabo de unas 4 semanas.
- 59. Diagnóstico de la vasculitis asociada a inmunoglobulina A (IgAV) Biopsia de lesiones cutáneas con inmunofluorescencia directa Depósitos inmunes con predominio de IgA1, que afectan a pequeños vasos en la piel y el tubo digestivo y causan con frecuencia artritis. La IgAV también se asocia con glomerulonefritis indistinguible de la nefropatía por IgA. Análisis de orina; la presencia de hematuria, proteinuria y cilindros eritrocíticos indica afectación renal. Hemograma completo y pruebas de función renal. Si hay deterioro de la función renal, la biopsia renal puede ayudar a definir el pronóstico.
- 60. Tratamiento de la vasculitis asociada a inmunoglobulina A (IgAV) Para adultos, medidas sintomáticas y corticosteroides con o sin un inmunosupresor En los niños, tratamiento sintomático para control el dolor. En los adultos 50 mg por vía oral 1 vez al día En caso de afectación renal grave metilprednisolona en pulsos IV seguida de prednisona por vía oral y un inmunosupresor (micofenolato de mofetilo, azatioprina, rituximab o ciclofosfamida)
- 61. VASCULITIS URTICARIAL HIPOCOMPLEMENTÉMICA ANTI c1q Es una vasculitis de pequeños vasos poco frecuente mediada por inmunocomplejos caracterizada por urticaria e hipocomplementemia (bajos niveles de C3, C4 y/o C1q), y generalmente asociada a autoanticuerpos anti-C1q circulantes. https://www.orpha.net/
- 62. VASCULITIS URTICARIAL HIPOCLOMPLEMENTEMICA ANTI c1q - Epidemiología La prevalencia es desconocida, pero se ha descrito menos de 500 casos en la literatura. Las mujeres se ven afectadas con mayor frecuencia que los varones (proporción mujer:hombre de 8:1). https://www.orpha.net/
- 63. SIGNOS Y SÍNTOMAS - Quinta década de la vida - Erupciones de urticaria generalizadas tronco, extremidades proximales y cara que a menudo se asocian con picazón o dolor y que persisten durante más de 24 horas, con hiperpigmentación después de la resolución. - Tos, disnea, derrame pleural y pericárdico y enfisema, con enfermedad pulmonar obstructiva crónica descrita en menos del 20% de los pacientes. - La enfermedad renal en el 20-30% de los pacientes es leve, con proteinuria y hematuria causada por la glomerulonefritis. - Síntomas digestivos (dolor abdominal, náuseas, diarrea, vómitos) - Musculoesqueléticas (artritis y artralgia transitoria que afecta a las manos, codos, rodillas, tobillos y pies) - Ocular (episcleritis, uveítis y conjuntivitis). https://www.orpha.net/
- 64. Etiología Asociados a mutaciones de DNASE1L3 (3p14.3). Se cree que los autoanticuerpos anti-C1q están implicados en la patogénesis del trastorno. https://www.orpha.net/
- 65. DIAGNÓSTICO Criterio mayor (urticaria recurrente durante >3 meses e hipocomplementemia) Criterio Menor (vasculitis leucocitoclástica en la biopsia, artralgia y artritis, inflamación ocular, dolor abdominal, glomerulonefritis y autoanticuerpos anti-C1q positivos). 1 criterio mayor más 2 criterios menores El patrón de herencia para los casos familiares asociados a DNASE1L3 es autosómico recesivo. Se recomienda proporcionar consejo genético a las familias de riesgo. https://www.orpha.net/
- 66. TRATAMIENTO Colchicina, disulona o hidroxicloroquina hasta terapias más agresivas, como esteroides y agentes inmunosupresores. Enfermedad cutánea y artralgias, pero sin afectación orgánica importante, colchicina, hidroxicloroquina o dapsona. Afectación orgánica importante, como la glomerulonefritis, dosis elevadas de corticosteroides y agentes citotóxicos similares a los empleados en el tratamiento del LES activo. La respuesta terapéutica suele ir acompañada de una disminución del título de anti-C1q circulante y de la normalización de los niveles de C3 y C4. https://www.orpha.net/
- 67. Pronóstico Influenciado principalmente por la gravedad de la enfermedad pulmonar, cardíaca y renal asociada. La enfermedad pulmonar es la principal causa de muerte. El edema laríngeo agudo puede poner en peligro la vida. https://www.orpha.net/
- 68. VASCULITIS DE VASO VARIABLE Enfermedad de Behçet Síndrome de Cogan https://www.orpha.net/
- 69. Enfermedad de Behçet Es una vasculitis multisistémica crónica y recurrente caracterizada por lesiones mucocutáneas, así como por manifestaciones articulares, vasculares, oculares y del sistema nervioso central. https://www.orpha.net/
- 70. Enfermedad de Behçet Epidemiología Mayor frecuencia en poblaciones de la Ruta de la Seda, con una mayor prevalencia registrada en Turquía de >1/1.000, respecto a 1/10.000 en Japón. https://www.orpha.net/
- 71. Enfermedad de Behçet Signos y Sintomas - Frecuente en adultos, episodios recurrentes consisten en aftas orales redondeadas con bordes elevados y eritematosos (1-3 cm de diámetro) pueden ir acompañados de aftas genitales (>50% de los casos) - Rasgos cutáneos pueden incluir pseudofoliculitis y eritema nodoso. - Trastornos oculares (uveítis posterior, vasculitis retiniana) en alrededor del 50% - Las artralgias y/o artritis simétrica no erosiva (45%) y pueden ser el síntoma inicial. - La vasculitis puede producir trombosis en venas fémoro-iliaca, cava superior e inferior y cerebral. Las trombosis y aneurismas arteriales afectan a los vasos pulmonares. - (neuro-Behçet) (>20%) cefalea, signos piramidales con hemiparesia, daños en los nervios craneales, meningitis, cambios conductuales y disfunción del esfínter. - Las lesiones aftoides y/o ulcerativas pueden afectar a la totalidad del tracto digestivo, pero fundamentalmente al ileocecal y al colon ascendente, pudiendo dar lugar a hemorragias y perforaciones. https://www.orpha.net/
- 72. Enfermedad de Behçet Etiologia Origen desconocido Predisposición genética en la EB puede favorecer que ciertas infecciones (en particular por Streptococcus sanguis) y/o agresiones ambientales El antígeno HLA B5101 está asociado a la EB en el 50-70% de los pacientes. La activación NF-kB y los niveles aberrantes de citoquinas (IL-6, TNF-a, IL-8, IL-12, IL-17 y IL-21) han sido implicados en la patogénesis de la EB. https://www.orpha.net/
- 73. Enfermedad de Behçet Diagnóstico Se basa en los criterios de clasificación internacional, que son sensibles y específicos. - La presencia de ulceración bucal recidivante (al menos 3 veces durante 12 meses; 2 puntos), - ulceración genital (2 puntos), - uveítis (2 puntos), - lesiones cutáneas (1 punto), - manifestaciones cardiovasculares (1 punto), - manifestaciones neurológicas (1 punto) y o reacción de patergia (1 punto). El diagnóstico se establece con una puntuación de ≥4 puntos. https://www.orpha.net/
- 74. Enfermedad de Behçet Diagnóstico Afectación visceral grave aislada (como trombosis de la vena cava, cerebral y/o subhepática, aneurismas pulmonares, afectación neurológica y/o vasculitis retiniana) Los antecedentes familiares de EB también incrementan la probabilidad de diagnóstico. La búsqueda del haplotipo HLA-B51 no es un elemento de diagnóstico sólido, pero puede resultar útil en situaciones de incertidumbre diagnóstica. https://www.orpha.net/
- 75. Enfermedad de Behçet Tratamiento Lorem ipsum Dolor nec Ipsum dolor amet dolor La colchicina alivia los síntomas mucocutáneos. 1 Esteroides son la base del tratamiento. Apremilast ha sido aprobado recientemente para la ulceración oral refractaria en la EB. 2 Inmunosupresores (como azatioprina, ciclofosfamida, metotrexato o 3 micofenolato de mofetilo) 4 El anti-TNF como el interferón alfa (2a o 2b) uveítis grave 5 Antiagregantes o anticoagulantes se utilizan en casos de trombosis vascular. https://www.orpha.net/
- 76. Enfermedad de Behçet Pronóstico Pronóstico es grave debido a la afectación ocular que puede producir ceguera, el riesgo de ruptura arterial es letal y los síntomas neurológicos pueden causar encefalopatía, lo que puede ocasionar al paciente gran invalidez. El cuidado oftalmológico intensivo junto con un tratamiento inmunosupresor ha demostrado reducir en gran medida la morbilidad. https://www.orpha.net/
- 77. Síndrome de Cogan Es un trastorno inflamatorio/ autoinmune poco frecuente de origen desconocido caracterizado por queratitis intersticial (QI) y disfunción audiovestibular. https://www.orpha.net/
- 78. Síndrome de Cogan Epidemiología La prevalencia del síndrome de Cogan (SC) es desconocida. Hasta la fecha, se han descrito 300 casos, aproximadamente, principalmente en individuos de raza caucásica sin predilección por sexo. https://www.orpha.net/
- 79. Sindrome de Cogan Signos y Sintomas Adultos jóvenes de inicio entre los 20 y 30 años, ocasionalmente a niños. Síntomas cocleovestibulares con pérdida auditiva neurosensorial uni- o bilateral, vértigo y tinnitus. El intervalo entre el inicio de la afectación ocular y audiovestibular suele ser inferior a los 2 años. Síntomas audiovestibulares distintos a los de la enfermedad de Menière, o cuando la latencia entre la afectación de estos dos órganos es superior a los 2 años. Síntomas generalizados tales como fiebre, cefaleas, pérdida de peso y/o presencia de signos de afectación orgánica, principalmente del sistema cardiovascular (aortitis, insuficiencia aórtica, insuficiencia cardíaca congestiva, fenómeno de Raynaud), Neurológico (neuropatía periférica, meningitis, hemiparesia o hemiplejía debido a un accidente cerebrovascular y afasia por un accidente isquémico transitorio) y gastrointestinal (diarrea, melena y dolor abdominal).
- 80. Síndrome de Cogan Etiología https://www.orpha.net/ Autoinmune
- 81. Diagnóstico El diagnóstico es fundamentalmente clínico por exclusión de infecciones (en primer lugar la sífilis y la enfermedad de Lyme) con una buena respuesta al tratamiento con corticosteroides. Audiograma, y las pruebas de imagen pueden ser útiles para respaldar el diagnóstico y excluir otras posibles etiologías. Diagnóstico diferencial Sífilis, la enfermedad de Menière y la enfermedad de Lyme, la sarcoidosis, la tuberculosis, la poliarteritis nodosa, la granulomatosis con poliangeítis y la arteritis de Takayasu. https://www.orpha.net/
- 82. Tratamiento Corticosteroides piedra angular Afectación ocular aislada y leve glucocorticoides tópicos asociados a fármacos ciclopléjicos Afectación ocular más grave corticosteroides sistémicos Prevención para discapacidad auditiva y las manifestaciones sistémicas (1-1.5mg/kg de prednisona al día) de 2 ó 3 semanas. Pacientes con enfermedad refractaria considerar un tratamiento (metotrexato, ciclofosfamida, azatioprina o ciclosporina A) Tratamiento con un inhibidor del factor de necrosis tumoral alfa (Infliximab) permitiendo una disminución gradual de la administración de corticosteroides, El uso temprano de infliximab como terapia de primera línea en casos graves parece ser aún más efectivo. El implante coclear es una estrategia quirúrgica de rescate valiosa en los casos de pérdida auditiva neurosensorial grave que no responde al régimen terapéutico inmunosupresor intensivo. https://www.orpha.net/
- 83. Pronóstico El pronóstico se relaciona principalmente con el riesgo de sordera permanente y con las complicaciones cardiovasculares, especialmente la insuficiencia aórtica. La afectación visceral grave o el fallecimiento asociado a la afectación cardiaca son infrecuentes. https://www.orpha.net/