
Recomendados
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Fibrosis qistica y ausencia de vasos deferentes clase basica
Similar a Fibrosis qistica y ausencia de vasos deferentes clase basica (20)
Más de braguetin
Fibrosis qistica y ausencia de vasos deferentes clase basica
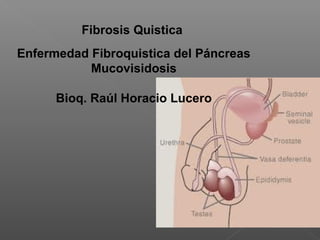
- 1. Fibrosis Quistica Enfermedad Fibroquistica del Páncreas Mucovisidosis Bioq. Raúl Horacio Lucero
- 2. Presentación Clínica Insuficiencia pancreática exócrina, con sus consecuencias nutricionales. Enfermedad pulmonar crónica (infecciónes recurrentes) Principal causa de morbilidad y mortalidad. Alteración de los valores de cloruro y sodio en el sudor. El 95% de los varones son estériles por ausencia bilateral congénita de vasos deferentes. Complicaciones más frecuentes íleo meconial (15%). Cirrosis hepática. Pólipos nasales. Síndrome de obstrucción intestinal distal Prolapso rectal. Edemas y hematomas (generalmente en neonatos)
- 3. Defecto Básico Mutaciones en el gen CFTR (Regulador de la Conductancia Transmembrana de la Fibrosis Quística) Codifica una proteína involucrada en el transporte de iones a través de la membrana celular. Genética Enfermedad monogénica. Es la enfermedad autosómica recesiva más frecuente en la población blanca. Probabilidad de tener un hijo afectado en una pareja de portadores es del 25% Incidencia de 1 en 2500 nacidos vivos. Frecuencia de portadores de 1 en 25. Heterogeneidad alelica asociada a heterogeneidad clínica
- 4. Características Moleculares Gen CFTR Proteína CFTR Regulador de la Conductancia Glicoproteína integral de membrana Transmembrana de la Fibrosis Quística. (1480 aa). Canal de Cloro regulado por Localización en el cromosoma 7. AMPcíclico. Mapea en 7q 31. Dos motivos repetidos: con un dominio transmembrana (TM1 y 2), Región codificante: 250 Kb. un dominio de unión a nucleótidos (NBF1 y 2) y un dominio Constituido por 27 exones. citoplasmático con función reguladora (R).
- 5. CFTR
- 6. Mutaciones del gen CFTR ∆F508: Deleción de tres pares de bases en el exón 10, que implica una pérdida de una fenilalanina en la posición 508 del péptido. Esta presente en el 67% de los cromosomas provenientes de afectados, a nivel mundial. Hay grandes variaciones geográficas y étnicas. Es la mutación más frecuente en todas las poblaciones a excepción de los Judios Ashkenazis (W1282X). Presenta alta frecuencia en el Norte de Europa (70%-80%), y más baja en el Sur de Europa (50%-55%). No se encontraron grandes deleciones. Más de 1500 mutaciones asociadas a la enfermedad Alrededor de 200 polimorfismos. Algunas mutaciones tienen una alta frecuencia en grupos étnicos específicos.
- 7. Espectro de mutaciones En la actualidad, se han identificado más de 1.500 cambios (mutaciones y polimorfismos) en el gen CFTR. La mutación más común a nivel mundial es una deleción de fenilalanina en posición 508 (p.F508del, sigla anterior ∆F508). Sin embargo, las mutaciones comunes (p.F508del, p.G542X, p.G551D, p.N1303K, p.W1282X) han mostrado diferentes frecuencias entre distintos grupos étnicos y localizaciones geográficas. La mayoría de las restantes mutaciones son menos frecuentes y generalmente se limitan a una región geográfica o a la aparición familiar exclusiva. Las mutaciones que provocan el cambio de un aminoácido por otro (en inglés "missense") representan un 41% de las mutaciones asociadas a enfermedad; las que provocan un codón de terminación prematuro (en inglés "nonsense"), un 10%; las inserciones o deleciones que causan un cambio en el marco de lectura (en inglés "frameshift"), un 18% y aquellas que alteran nucleótidos conservados en sitios de unión intrón-exón y afectan el procesamiento del pre- ARNm (en inglés "splicing"), un 13%. Existen 3% de rearreglos genómicos (grandes deleciones o inserciones) y otro 1% que afecta la zona promotora. Se han publicado datos sobre alrededor de un 14% de cambios neutros que no ocasionan enfermedad denominados polimorfismos.
- 8. Características Clínicas de pacientes Fibroquísticos Fenotipo Severo Fenotipo Moderado Edad de diagnóstico Temprana Tardía (1 a 2 años) (> de 10 años) Función Pancreática IP SP Estado Nutricional Pobre Bueno Ileo Meconial Alta Incidencia (15%) No Cl en sudor (> 80 mEq./l) 40-80 mEq./l Fertilidad en hombres CBAVD CBAVD Función pulmonar Variable Variable Mutaciones asociadas 2 alelos severos 1 alelo moderado
- 9. Tipos de Mutaciones de acuerdo al Fenotipo Mutaciones Severas Mutaciones Moderadas ∆F508 R117H G542X A455E G551D 3849 + 10 Kb C → T R553X R334W 621 + 1 G → A R347P W1282X R347H N1303K R3529 1717 − 1 G → A 2789 + 5 G → A
- 10. Clases de Mutaciones del gen CFTR G542X W1282X ∆F508 G551D R117H N1303k R334W 3849+10KbC→T R1162X G551S S549R R347P ALELOS 5T R553X S1225 621+1G→T ∆I507 A455E
- 11. Polimorfismos Pueden influenciar en el fenotipo Más estudiado en el CFTR Alelos T (intrón 8) El ejemplo más estudiado, con la mutación R117H: R117H/∆F508 → 7T / 7T → CBAVD CBAVD R117H/∆F508 → 5T / 7T Test del Sudo R117H/∆F508 → 5T / 5T patológico o límite Enfermedad pulmonar leve a moderada Suficiencia pancreática exócrina R117H/∆F508 → 7T / 7T Mujeres No sintomáticas
- 12. Resumen Fenotipo pancreático fuertemente correlacionado con el genotipo. Mutaciones usualmente asociadas a IP y otras a SP. Insuficiencia Pancreática (IP) → severo Suficiencia Pancreática (SP) → moderado El diagnóstico temprano y las diferencias en los tratamientos serían los responsables de la variabilidad en la severidad de la enfermedad pulmonar, y de la ocurrencia de las complicaciones más comunes de la Fibrosis Quística. Existen otros factores genéticos, dentro y fuera del gen CFTR, así como también factores ambientales.
- 13. APLICACIONES DEL DIAGNOSTICO MOLECULAR Asesoramiento Genético. Familias con historia previa de FQ. Cosanguineidad. Diagnóstico de la enfermedad. Individuos con síntomas clínicos sugestivos de FQ en los que os métodos clásicos no son concluyentes. Diagnóstico prenatal cuando por ultrasonido se detecta íleo meconial, bolo ecogénico o bolo obstructivo. Screening positivo. Infertilidad masculina por ausencia bilateral de vasos deferentes. (se estudia también a la pareja para los cálculos de riesgo). Ovo donación y espermodonación. Estudios de Correlación Genotipo/ Fenotipo Terapéutica ?
- 14. DIFICULTADES PARA EL DIAGNOSTICO MOLECULAR DE LA FIBROSIS QUISTICA Gran espectro de mutaciones asociadas a la enfermedad. Las diferentes mutaciones presentan grandes variaciones en sus frecuencias en las diferentes poblaciones. Grandes variaciones en el número de mutaciones para un análisis genotípico completo. Recomendación: Realizar estudios poblacionales para tener idea de la frecuencia de mutaciones y poder establecer el rango de confianza. Si existe historia de FQ en la familia, y no se determinó la mutación, realizar estudios de ligamiento genético. Quedan casos sin resolver. Métodos costosos y laboriosos.
- 15. Metodos de Diagnostico Molecular Metodos Directos Métodos Indirectos Uso de diferentes técnicas Marcadores extra o intra génicos. para detectar alteraciones en el ADN. No especifican la mutación. Son más complejos y laboriosos. Uso de Kits comerciales Búsqueda de nuevas mutaciones DGGE. SSCP. TGGE. Secuenciacion.
- 16. Ausencia Bilateral Congénita de Vasos Deferentes (CBAVD) Representa del 1 al 2% de la esterilidad masculina, y hasta el 25% de la azoospermia obstructiva global. Cuando está aislada es considerada como una entidad clínica particular, pero debido a que está presente en el 95% de los varones con Fibrosis Quística (FQ), Holsclaw propuso que los varones con CBAVD presentarían una forma leve de FQ. La hipótesis de Holsclaw fue corroborada años más tarde cuando fue posible identificar y clonar el gen causante del defecto básico de FQ detectándose mutaciones del CFTR en pacientes CBAVD. Es causada por el mal desarrollo de los conductos mesonéfricos generando agenesia o atresia del epidídimo, los vasos deferentes, y las vesículas seminales. Presenta un patrón de herencia autosómica recesiva. .
- 17. La severidad de las manifestaciones clínicas de la FQ, está relacionada con las diferentes clases de mutaciones del gen CFTR. La CBAVD por su desarrollo, sería una forma de presentación FQ ubicada en el extremo leve de un gradiente de expresión. Los pacientes CBAVD presentan las mismas mutaciones que los FQ pero con otra frecuencia y algunas variantes propias (IVS 8-5T). Se comprobó una asociación entre la presencia de mutaciones y sintomatología compatible con FQ en pacientes con CBAVD. Si bien los pacientes CBAVD tendrían una forma preponderantemente genital de FQ, en algunos ciertos síntomas referidos exigen profundizar los estudios pulmonares y digestivos propios de la patología FQ.
- 18. Diagnóstico de CBAVD Ausencia de vasos deferentes, epidídimo y/o vesículas seminales en la palpación y/o ultrasonografía. Análisis bioquímico del semen : Azoospermia. Bajo volumen (< 0.5 cc) pH< 7 Alta concentración de ácido cítrico Ausencia de células redondas Peroxidasa negativo. Baja concentración de fructosa.
- 19. Asesoramiento Los pacientes con CBAVD tienen espermatogénesis normal. Actualmente es posible el tratamiento de su infertilidad. Si la CBAVD está asociada con mutaciones en el CFTR, estos pacientes tienen riesgo aumentado de tener hijos FQ o CBAVD, ya que tienen el 100% de probabilidad detransmitir una copia alterada del gen. La mujer debe ser estudiadas para las mutaciones FQ .
- 20. Zona regulatoria 250.000 pb (27 Exones) F508 Pérdida de Fenilalanina en posición 508 Deleción de 3 Nt (CTT) entre los Nt 1652-1655
- 21. PCR(ΔF508) Sangre Extracción de Alelo específica periférica ADN NORMAL Electroforesis MUTADO HOMOCIGOTA HETEROCIGOTA
- 22. FREQUENT CYSTIC FIBROSIS MUTATIONS Name Relative frequency Mutation Consequence F508 67.2 Deletion de 3pb, Ex 10 Del Phe G542X 3.4 G T, Ex 11 Gly Stop W128X 2.1 G A, Ex 20 Trp Stop
- 23. Pacientes Hosp. Pediátrico (Rcia.) F1 (4 a) 02-05-03 HOMO. M P/P F 31% F2 (2 a) 19-05-03 HOMO. N E C TS +- F3 (1 a) 04-06-03 HETE. M P/P TS +- F 23% F4 (7 a) 26-06-03 HETE. M CONTROL F5 (21 a) 20-08-03 HETE M mamá 46% F6 (31 a) 20-08-03 HETE. M papá F7 (7 a) 26-08-03 HETE. M P/P F8 (1 a) 22-09-03 HETE. M PUL. F9 (8 m) 29-09-03 HOMO. M PUL. TS + F10 (3m) 26-04-04 HOMO. M PUL. F11 (6 m) 30-04-04 HOMO. N F F12 (3 m) 18-04-04 HOMO. N TS + F13 (11 a) 01-07-04 HOMO. N TS +
- 24. PACIENTES ROSARIO FR1 (21 a) HETE. M IP. Hna. F. 13% FR2 (34 a) HETE. M IP FR3 HETE. M 20% FR4 HETE. M FR5 (19 a) HETE. M TS + P/P 67% FR6 HOMO. M FR7 (17 a) HOMO. M CONTROL P/P FR8 (12 a) HOMO. N IP TS + P/P FR9 (8 a) HETE. M IP Control FR10 (15 a) HETE. M IP TS+ Control FR11 (14 a) HOMO. M P/P TS+ FR12 (33 a)HF2 HETE. M P/P FR13 HETE. M FR14 HOMO. N FR15 HETE. M
- 26. Foto de corrida electroforética en gel de agarosa al 2%. De izquierda a derecha: w16d16,w17d17,w20d20,w20´d20´ ,mpm ,w21d21,w21´d21´,w-d- . Paciente w16d16: Homocigota mutado Paciente w17d17: Heterocigota mutado Paciente w20d20: Heterocigota mutado Duplicado paciente anterior w20´d20´: Heterocigota mutado Mpm: Marcador de Peso molecular Paciente w21d21: Homocigota normal Duplicado paciente anterior w21´d21´ : Homocigota normal w-d-: Control negativo