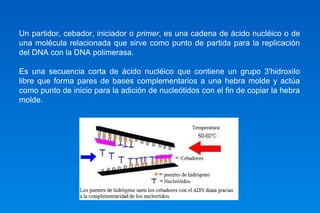
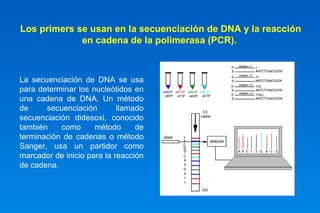
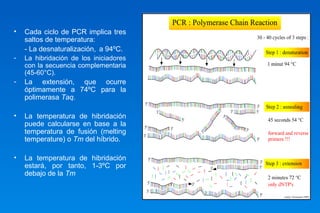
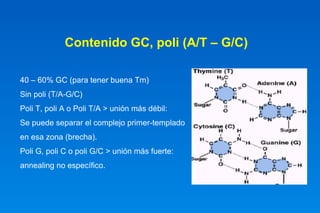
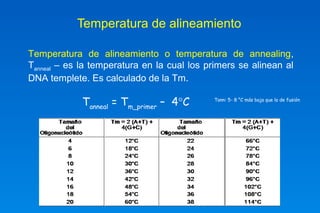
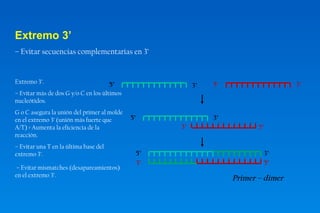
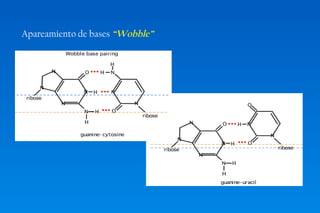
Este documento describe los principios básicos del diseño y construcción de iniciadores y sondas de DNA para ingeniería genética. Explica que los iniciadores son secuencias cortas de ácido nucleico que sirven como punto de inicio para la replicación del DNA. Luego detalla varios criterios importantes para el diseño de iniciadores como la longitud, contenido GC, temperatura de fusión, especificidad y evitar la formación de dímeros. También cubre el uso de iniciadores degenerados y la importancia de validar la especificidad de la secuencia del
































































































![Curso gen..[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cursogen-1-121031063927-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)
![molecular__diapositiva_30-60[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/moleculardiapositiva30-601-230817050758-b61eec83-thumbnail.jpg?width=640&height=640&fit=bounds)
